figures
Alexander Haglund
2022-11-10
Last updated: 2022-12-01
Checks: 7 0
Knit directory: SingleCellMR/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221110) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version d902176. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: analysis/downstream.nb.html
Ignored: analysis/figures.nb.html
Untracked files:
Untracked: analysis/DOWNSTREAM_RESULTS.Rmd
Untracked: analysis/eQTL_analysis.Rmd
Untracked: colossus/
Untracked: data/COLOC_MR_RESULTS/
Untracked: data/EXT_DATASETS/
Untracked: data/FIGURES/
Untracked: data/GWAS_STUDIES/
Untracked: data/MARKDOWN/
Untracked: data/METADATA/
Untracked: data/TABLES/
Untracked: data/derby.log
Untracked: data/eQTL_RESULTS/
Untracked: data/helper_files/
Untracked: data/logs/
Untracked: derby.log
Untracked: logs/
Unstaged changes:
Modified: analysis/_site.yml
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/figures.Rmd) and HTML
(docs/figures.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | d902176 | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 7389e87 | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 8db15db | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | dbf14b0 | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 7b58517 | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 0f7883c | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | ed4677c | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | bb50a67 | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 5587527 | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 10dd725 | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 0758ce5 | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | dd4ab87 | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 762caf0 | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 8f21a16 | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | a155fea | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | babc9f5 | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 6966b34 | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 3c46f4b | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 4b93157 | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | fd90855 | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 8a35089 | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 016f567 | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 4639838 | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 43e9b1f | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | ad8d30b | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 3c08408 | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 9aafc1c | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 3f6c35b | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | 2fd70c5 | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 4fcf3ea | Alexander Haglund | 2022-12-01 | Build site. |
| Rmd | eacb4eb | Alexander Haglund | 2022-12-01 | wflow_publish(c("analysis/figures.Rmd")) |
| html | 597c47b | Alexander Haglund | 2022-11-18 | Build site. |
| html | 8f071f3 | Alexander Haglund | 2022-11-18 | Build site. |
| Rmd | 38a3835 | Alexander Haglund | 2022-11-18 | wflow_publish(c("analysis/index.Rmd", "analysis/figures.Rmd")) |
libraries
library(ggplot2)
library(viridis)
library(ggsci)
library(dplyr)
library(cowplot)
library(grid)
library(tidyr)
suppressMessages(library(reshape))
color_pal=ggsci::pal_nejm("default")(8)
colorvec<-c(Astrocytes=color_pal[1],
Endothelial=color_pal[2],
Excitatory=color_pal[3],
Inhibitory=color_pal[4],Microglia=color_pal[5],
ODC=color_pal[6],OPC=color_pal[7],Pericytes=color_pal[8])FIGURE 1
coming soon
FIGURE 2
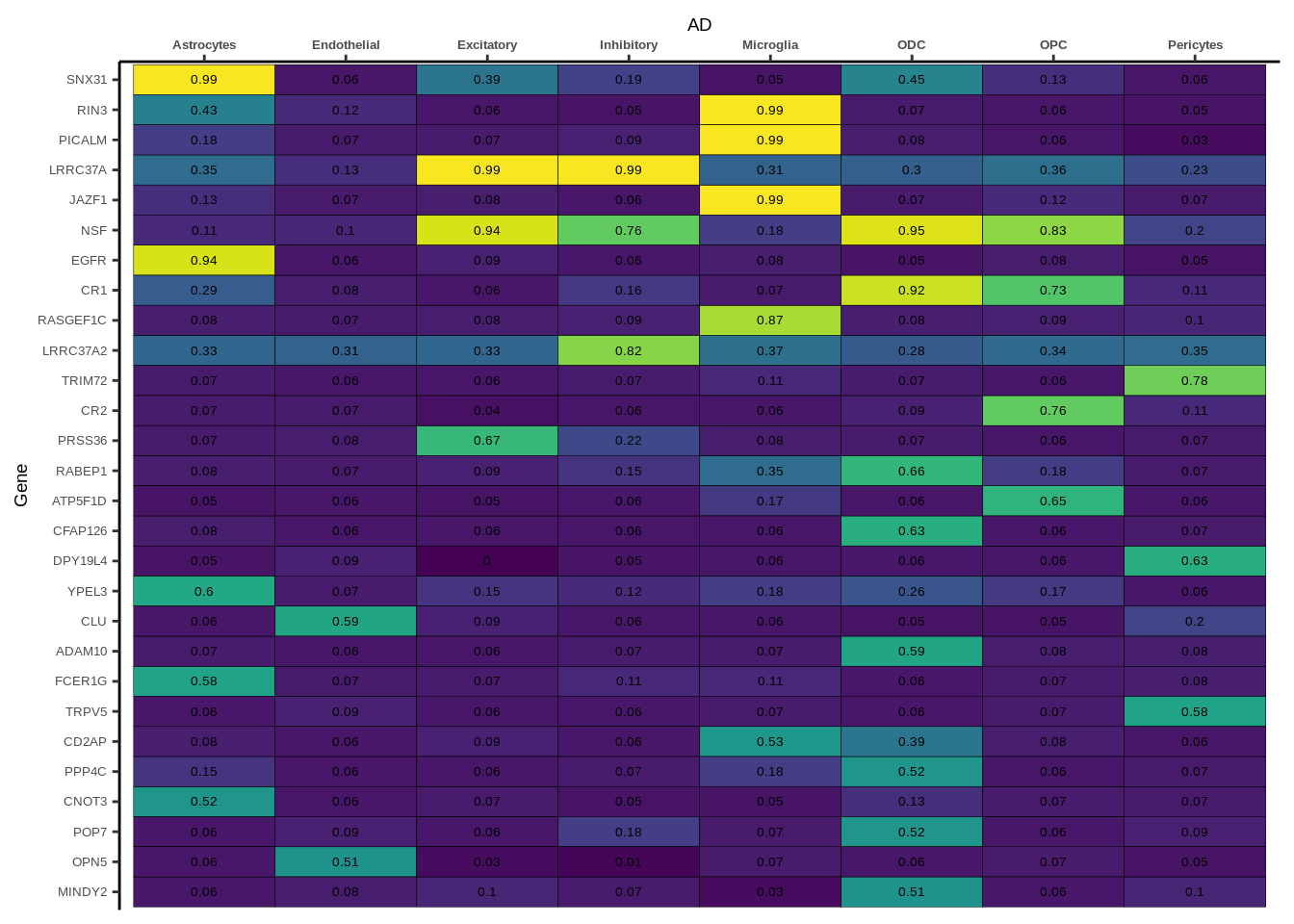
Figure 2a
coloc<-read.table("data/COLOC_MR_RESULTS/2022-10-25_FULL_COLOC_RES.txt")
x<-coloc[coloc$GWAS %in% "AD",]
genes<-x[x$PP.H4.abf>0.5,]$gene
x<-x[x$gene %in% genes,]
x$gene<-factor(x$gene,levels=unique(x$gene))
g<-ggplot(x,aes(x=celltype,y=gene,fill=PP.H4.abf))
g<-g+geom_tile(aes(fill=round(PP.H4.abf,2)),colour="black")+
geom_text(aes(label = round(PP.H4.abf, 2)),size=5*0.36,family="Helvetica")+
scale_fill_viridis()+
theme_classic()+
scale_y_discrete(limits=rev,expand = c(0, 0))+
scale_x_discrete(expand = c(0, 0),position="top")+
xlab("Alzheimer's Disease")
g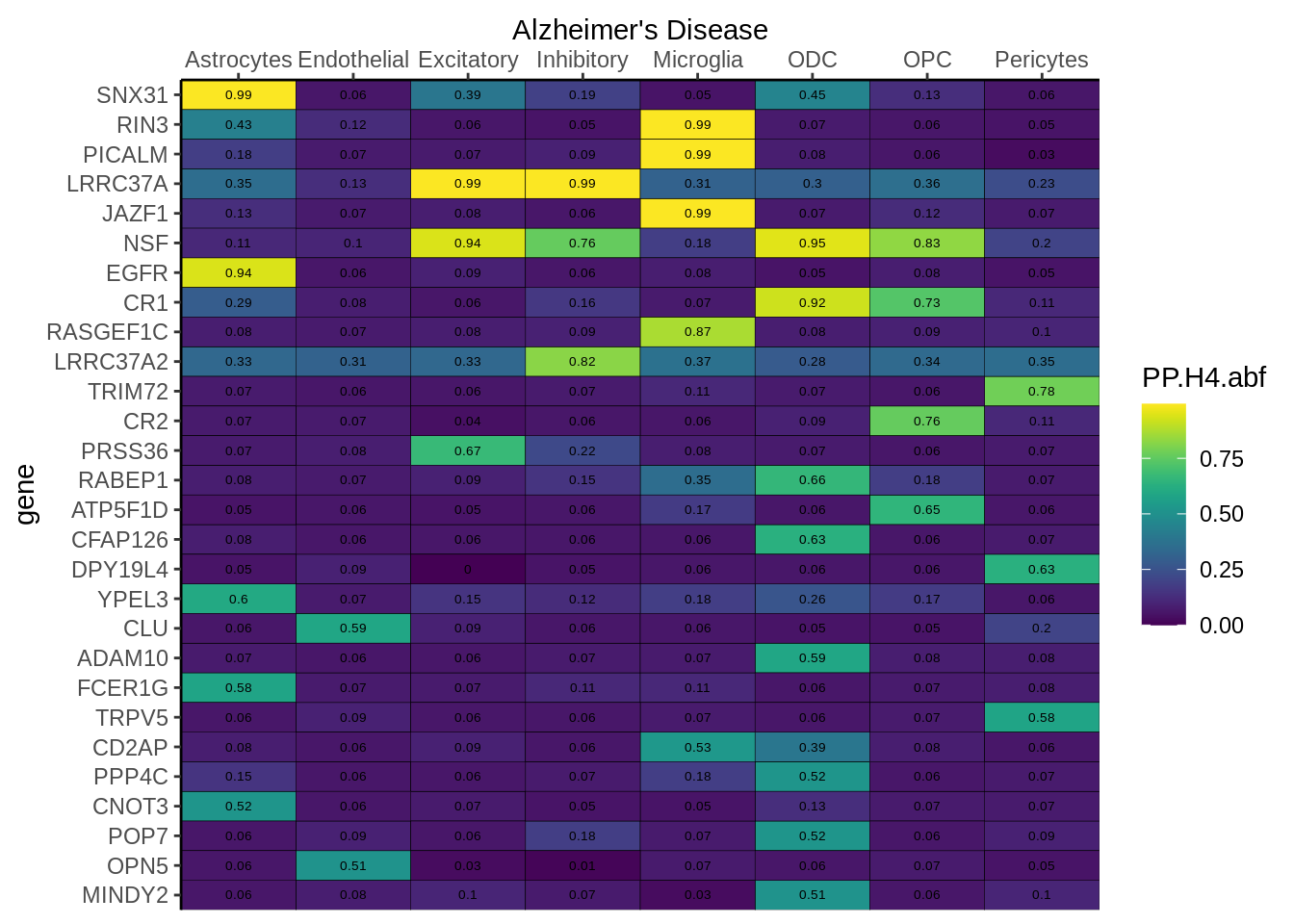
| Version | Author | Date |
|---|---|---|
| 8f071f3 | Alexander Haglund | 2022-11-18 |
Figure 2b
coming soon
Figure 2c
coming soon
Figure 2d
coloc_results<-read.table("data/COLOC_MR_RESULTS/2022-10-25_FULL_COLOC_RES.txt")
fig_dir<-"FIGURES/Figure_2/"
coloc_results<-coloc_results[coloc_results$PP.H4.abf>0.5,]
g<-ggplot(coloc_results,aes(y=GWAS))+geom_bar(color="black",fill="#166FA2")+
geom_text(aes(label=..count..),stat="count",hjust=-0.8,size=5/(14/5))+
labs(x="Number of colocalisations")+
theme_classic()+scale_x_continuous(limits=c(0,120),expand = c(0, 0))+
theme(text=element_text(family="Helvetica",face="bold"),
axis.text.x=element_text(size=5,face="bold"),
axis.text.y=element_text(size=5),
axis.title.x=element_text(size=5),axis.title.y=element_text(size=5))
g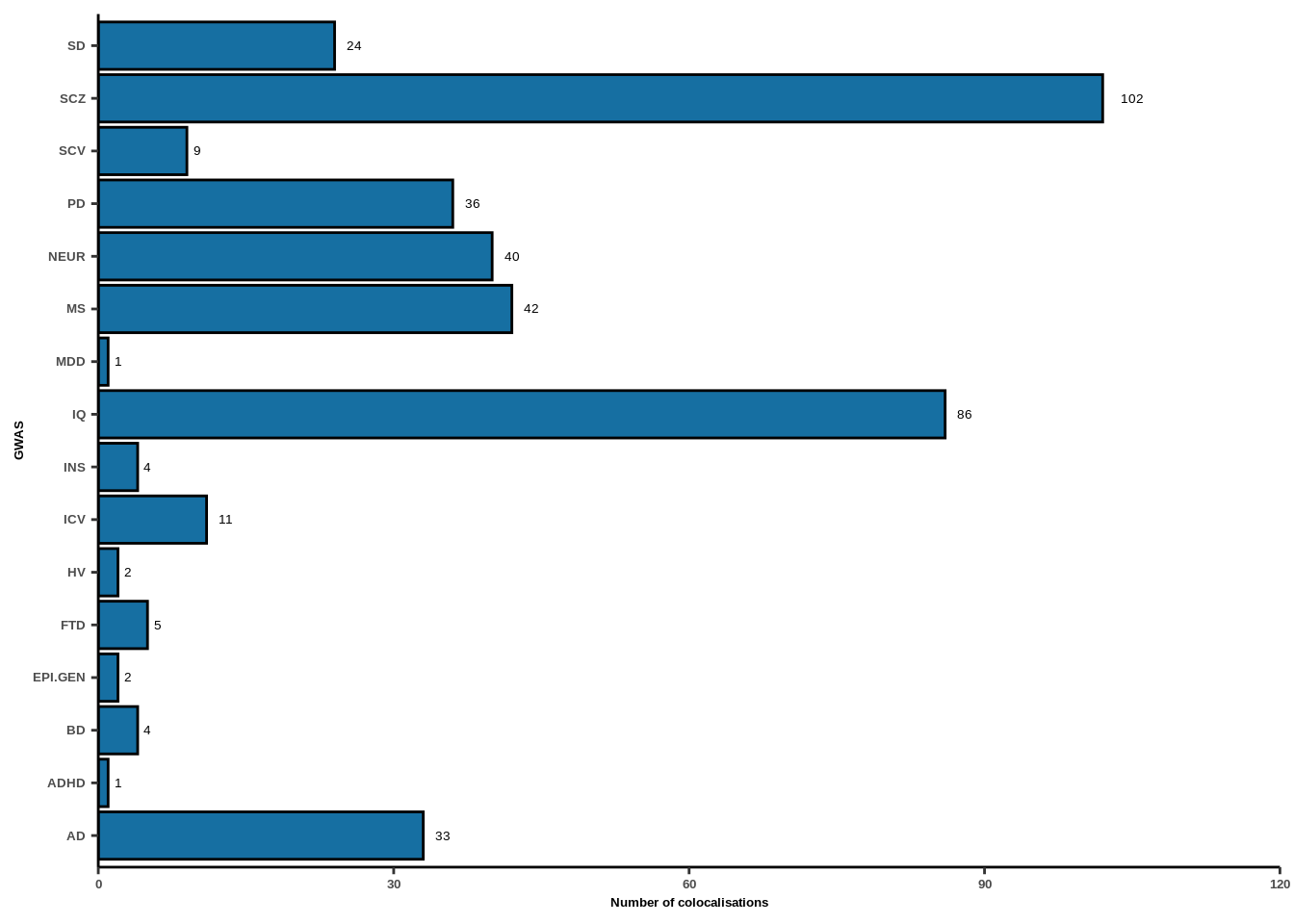
| Version | Author | Date |
|---|---|---|
| 3f6c35b | Alexander Haglund | 2022-12-01 |
Figure 2e
coloc<-read.table("data/COLOC_MR_RESULTS/2022-10-25_FULL_COLOC_RES.txt")
##rename SCV caudate for spacing
coloc_results$GWAS<-sapply(coloc_results$GWAS,function(x){
if(x=="SCV.CAUDATE"){
x<-"SCV"}
return(x)})
##filter to keep coloc hits
coloc_results_filtered<-coloc_results[coloc_results$PP.H4.abf>0.5,]
gwas_list<-unique(coloc_results_filtered$GWAS)
reslist<-list()
##count number of cell types per trait
for(i in 1:length(gwas_list)){
tmp<-coloc_results_filtered %>% filter(GWAS==gwas_list[i])
freq_table<-as.data.frame(table(tmp$celltype))
tmp$celltype_freq<-freq_table[match(tmp$celltype,freq_table$Var1),]$Freq
#scale by total number of colocs (otherwise larger gwases will have really large points)
# tmp$celltype_freq<-tmp$celltype_freq/nrow(tmp)
reslist[[i]]<-tmp
}
coloc_results_filtered<-as.data.frame(do.call(rbind,reslist))
color_pal=ggsci::pal_nejm("default")(8)
g<-ggplot(coloc_results_filtered,aes(y=GWAS,x=celltype,size=celltype_freq,fill=celltype))+
geom_point(pch=21)+
scale_size(range=c(1,3))+
# geom_text(aes(label=celltype_freq),size=5/(14/5),vjust=-1.1)+
theme_classic()+
scale_fill_manual(values=color_pal)+
theme(text=element_text(family="Helvetica",face="bold"),
axis.text.x=element_text(size=5,angle=45),
axis.text.y=element_text(size=5),
axis.title.x=element_text(size=5),axis.title.y=element_text(size=5),
legend.text=element_text(size=5),legend.title=element_text(size=5),
legend.spacing.y = unit(0.05, 'cm'),legend.position = "none")
g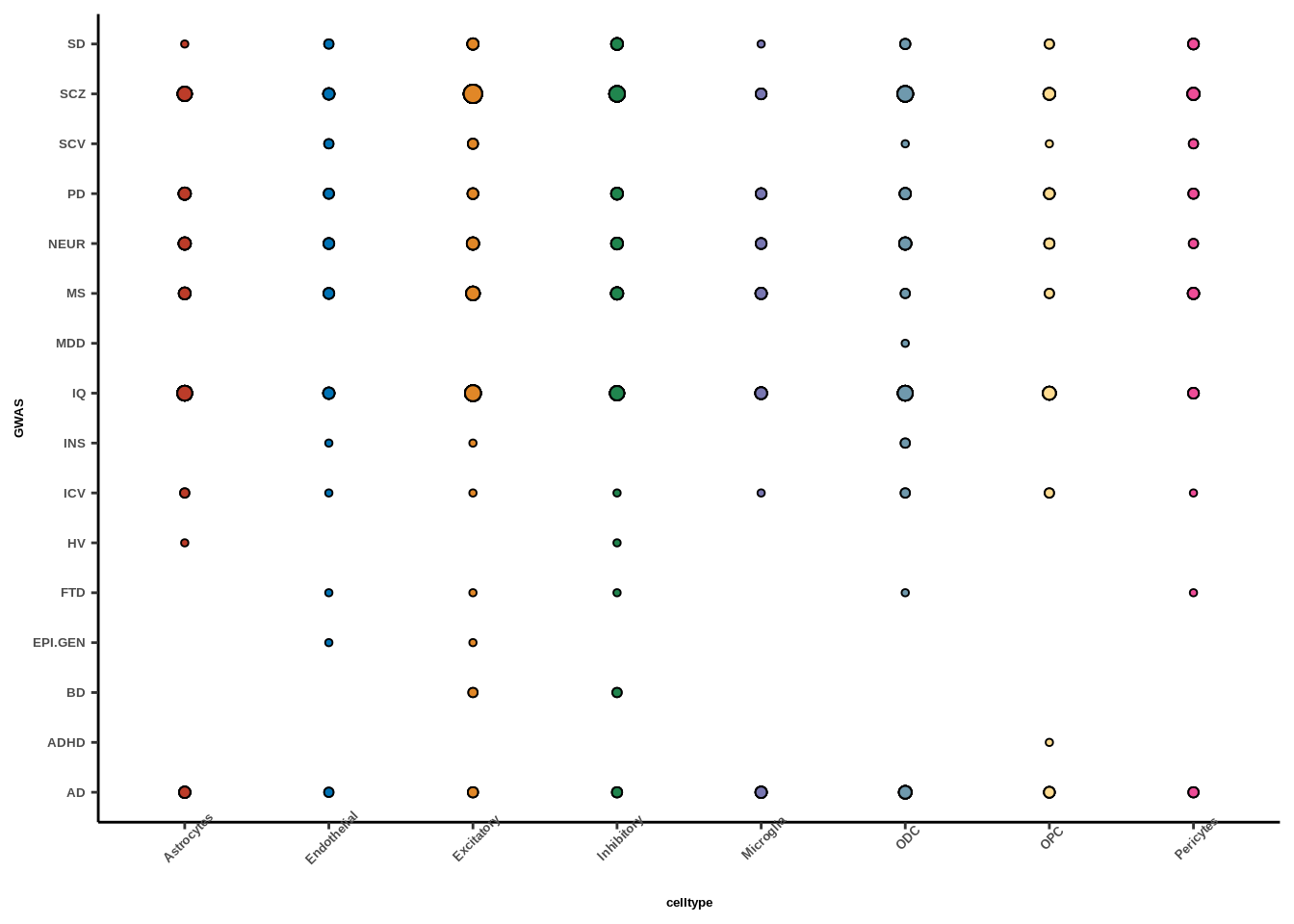
FIGURE 3
The code presented here is in step 5 (Epigenetic Intersection) of the downstream results section. The basic syntax is a geom_tile() function in ggplot with colours matching cell-types. More in data/MARKDOWN/helper_funcs.r
This is only for Fig 3.a - Fig 3.c and Fig 3.d were made using the UCSC track browser.
indir<-"data/EXT_DATASETS//RESULTS/"
fig_dir<-"FIGURES/Figure_3/"
g1<-readRDS(paste0(indir,"oligo_intersect_ggobject.rds"))
g2<-readRDS(paste0(indir,"Excneuron_intersect_ggobject.rds"))
g3<-readRDS(paste0(indir,"Inneuron_intersect_ggobject.rds"))
g4<-readRDS(paste0(indir,"microglia_intersect_ggobject.rds"))
g1<-g1+theme(legend.position = "none")
g2<-g2+theme(legend.position = "none")
g3<-g3+theme(legend.position = "none")
g4<-g4+theme(legend.position = "none")Oligo
g1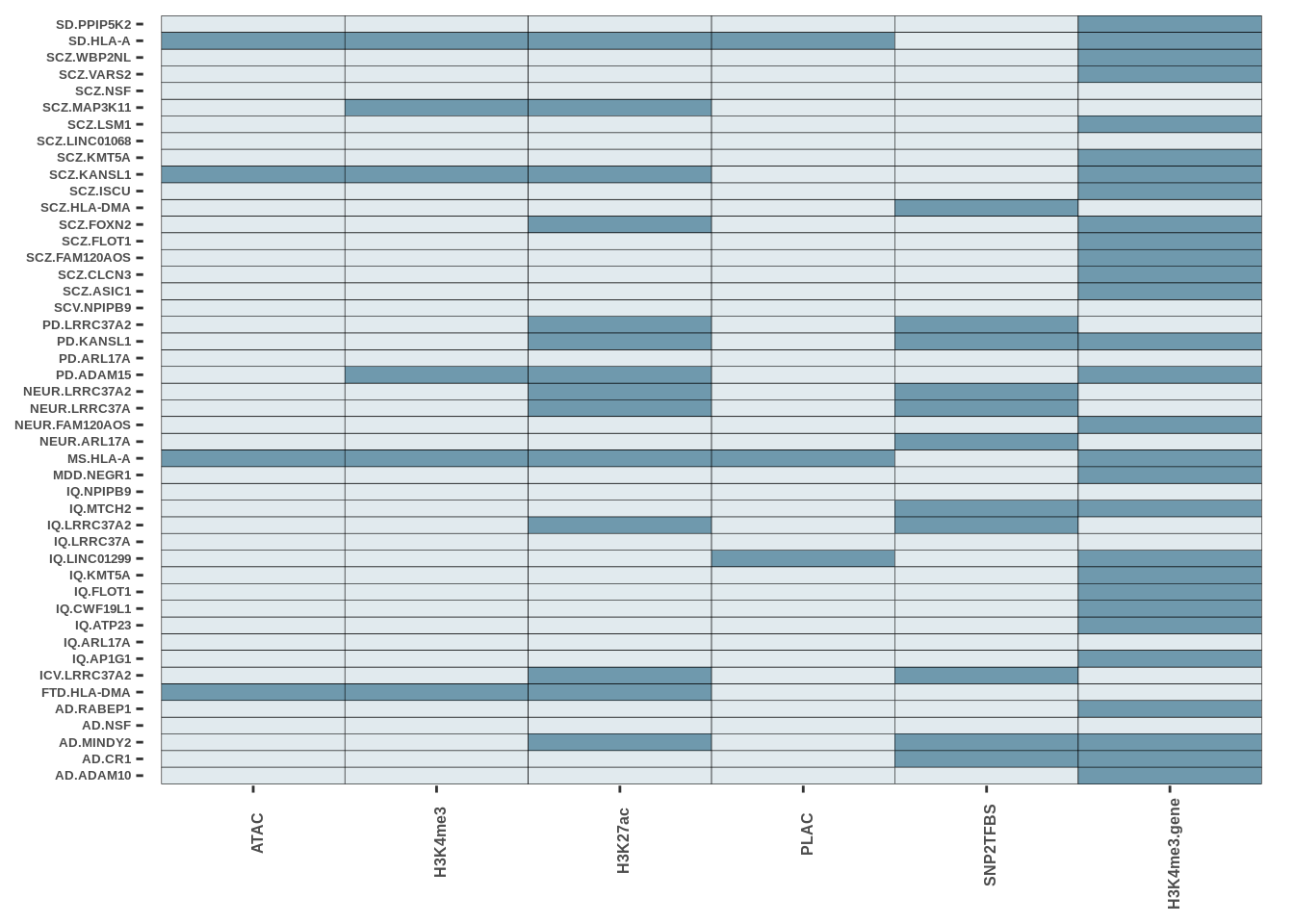
ExcNeur
g2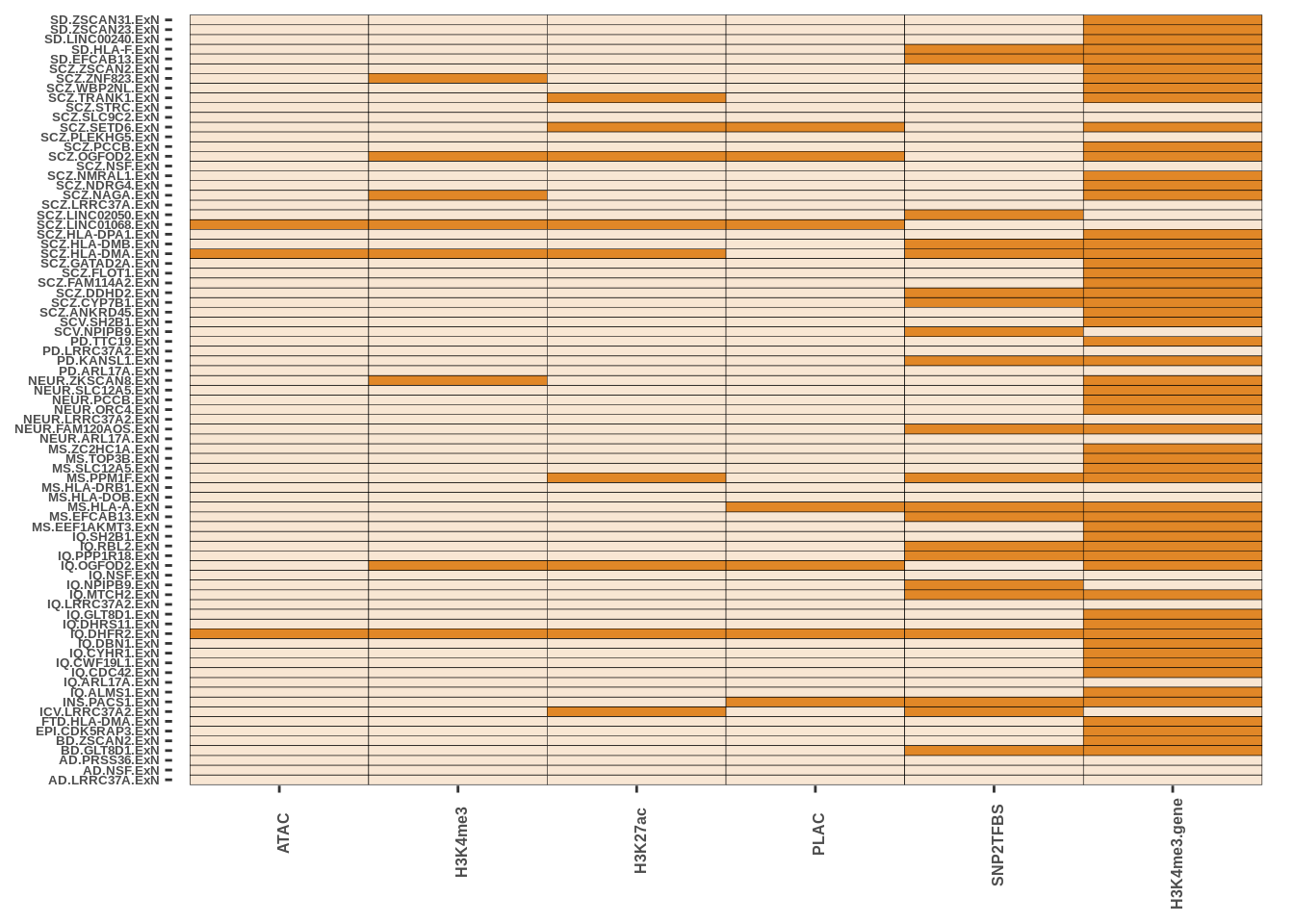
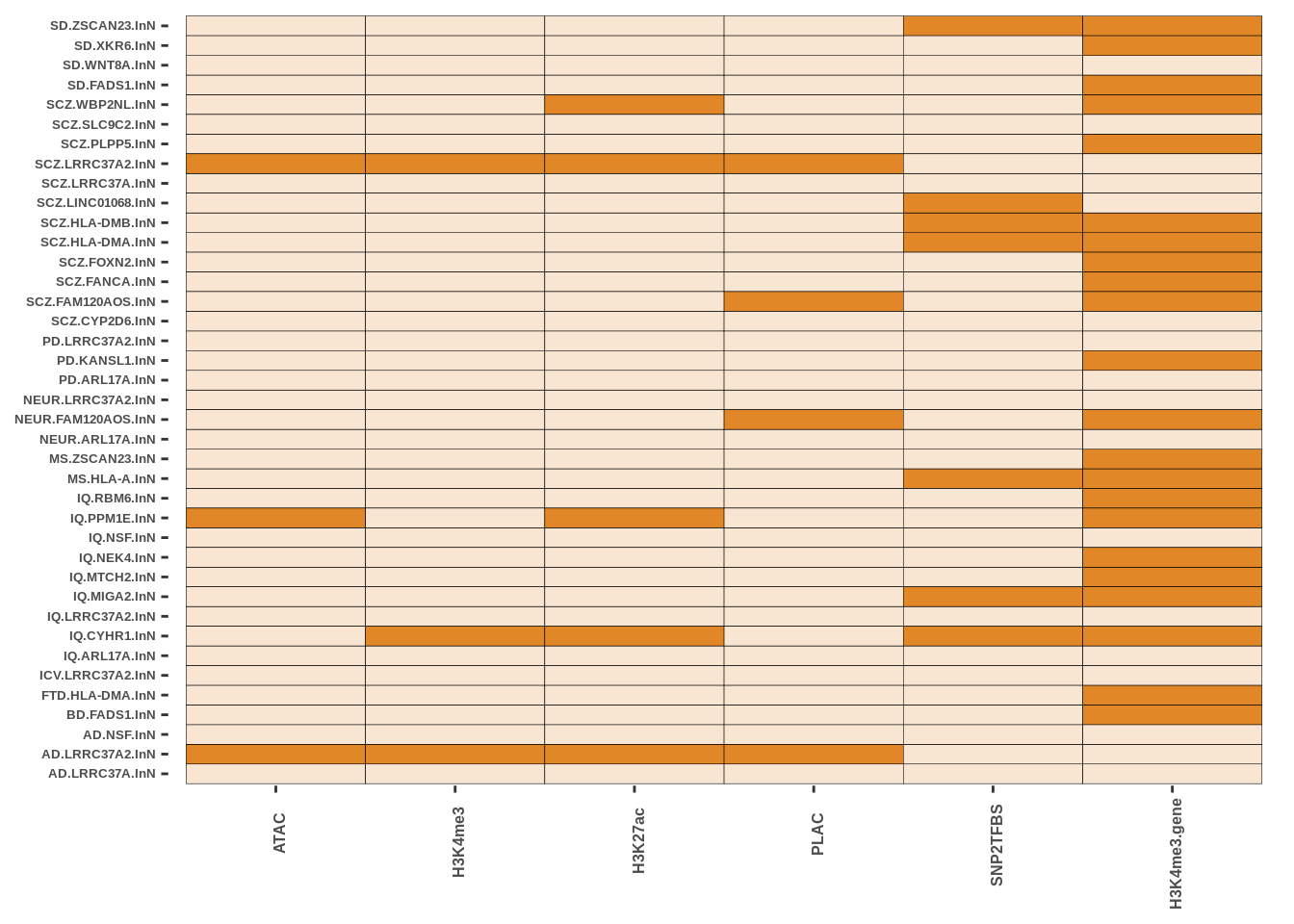
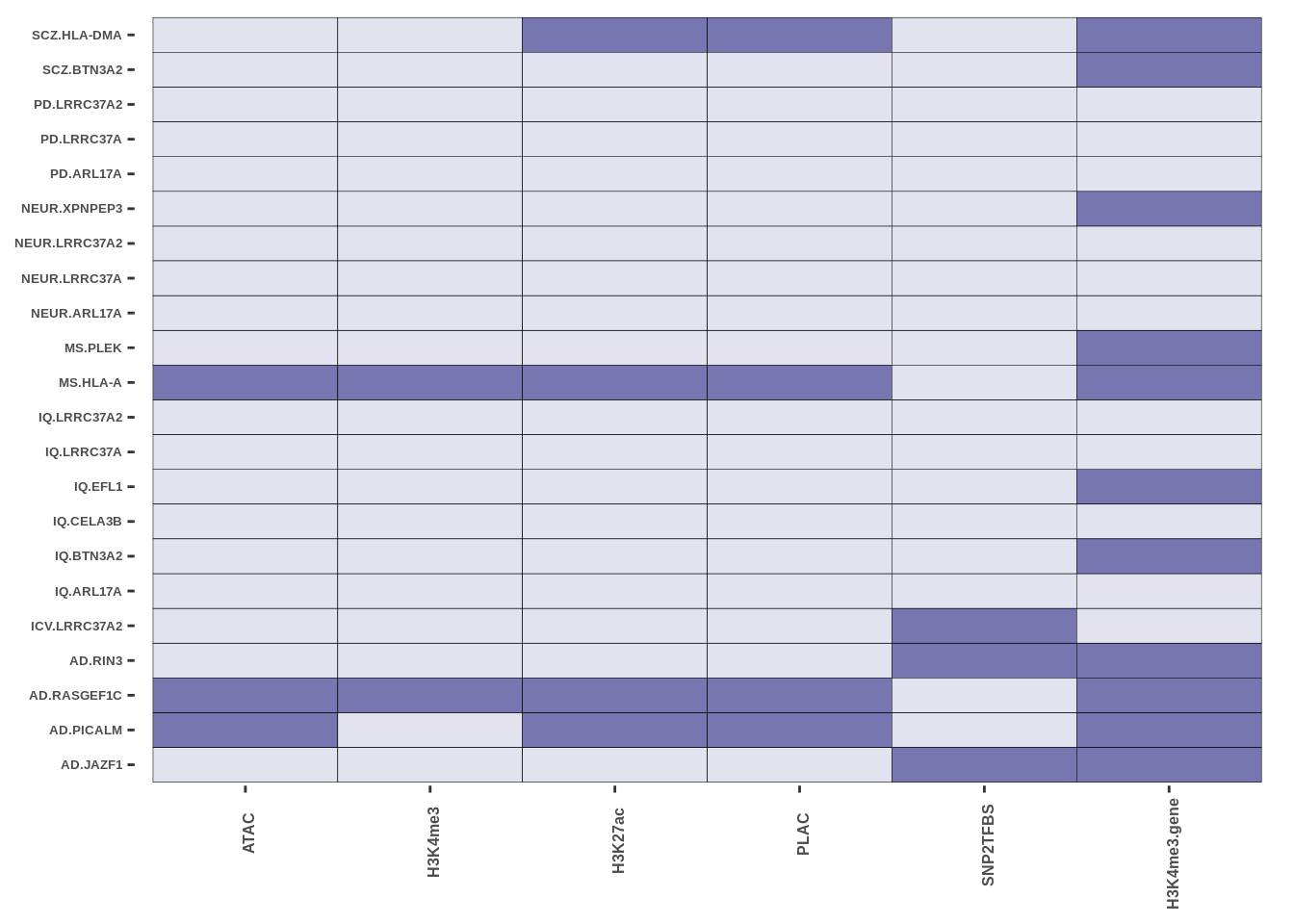
FIGURE 4
Figure 4a
bind data together
#bind data together
pqtl<-read.table("data/TABLES/pQTL_table.txt")
stitch<-read.table("data/TABLES//stitch_table.txt")
dgidb<-read.table("data/TABLES/dgidb_table.txt")
opentargets<-read.table("data/TABLES/OpenTargets_table.txt")
coloc<-read.table("data/COLOC_MR_RESULTS//2022-10-25_FULL_COLOC_RES.txt")
full<-read.table("data/COLOC_MR_RESULTS//2022-10-25_FULL_MR_RES.txt")
full<-full[full$IVW<0.05,]
full$trait_gene<-paste0(full$GWAS,"_",full$gene)
full$IVW_dir<-sapply(full$IVW_beta,function(x){
if(x>0){
return("positive")
}else{
return("negative")
}
})
direction_vector<-vector()
for(i in 1:nrow(full)){
tmp<-full[full$trait_gene %in% full$trait_gene[i],]
betas<-tmp$IVW_beta
if(all(betas>0)==TRUE){
dir<-"positive"
} else if(all(betas<0)==TRUE){
dir<-"negative"
}else{
dir<-"N/A"
}
direction_vector<-c(direction_vector,dir)
}
full$IVW_dir<-direction_vector
coloc$trait_gene_ct<-paste0(coloc$GWAS,"_",coloc$gene,"_",coloc$celltype)
trait_gene_ct<-paste0(pqtl$GWAS,"_",pqtl$gene,"_",pqtl$celltype)
coloc<-coloc[match(trait_gene_ct,coloc$trait_gene_ct),]
plot_df<-data.frame(gene_trait=paste0(pqtl$GWAS,".",pqtl$gene),
pQTL=pqtl$pQTL_hit,
STITCH=stitch$STITCH_intersect,
DGidb=dgidb$DGIDB_intersect,
OpenTargets=opentargets$OpenTargets_disease_hit,
coloc=coloc$PP.H4.abf,
celltype=pqtl$celltype,
IVW_dir=full$IVW_dir)prep plots
##intersection plot
plot_df1<-plot_df[,c("gene_trait","pQTL","STITCH","DGidb","OpenTargets")]
melt_df<-melt(plot_df1,id=c("gene_trait"))
ggplot_mainplot1<-ggplot(melt_df,aes(x=variable,y=gene_trait,fill=value))+geom_tile(color="black")+
scale_fill_manual(values=c("#c8c8c8","#A6CEE3","#1F78B4"))+scale_x_discrete(expand=c(0,0))
##direction
ivw_dir<-plot_df[,c("gene_trait","IVW_dir")]
ivw_dir$trait<-"gwas"
ivw_dir1<-ivw_dir
ggplot_ivwdir1<-ggplot(ivw_dir1,aes(y=gene_trait,x=trait,fill=IVW_dir,group=IVW_dir))+geom_tile(colour="black")+
scale_fill_manual(values = c("#c8c8c8","#F8766D","#00BFC4"),guide = guide_legend(reverse = TRUE))+
theme_classic()
##celltypes
celltype_bar<-plot_df[,c("gene_trait","celltype")]
celltype_bar1<-celltype_bar
test<-celltype_bar1 %>%
count(gene_trait,celltype,name="count") %>%
complete(gene_trait,celltype)
newvec<-vector()
for(i in 1:nrow(test)){
if(is.na(test$count[i])){
newvec<-c(newvec,NA)
}else{
newvec<-c(newvec,test$celltype[i])
}
}
test$new<-newvec
# melt_df<-melt(celltype_bar1,id=c("gene_trait"))
# df2 = celltype_bar1 %>% complete(gene_trait,celltype)
ggplot_celltype1<-ggplot(test,aes(x=celltype,y=gene_trait,fill=new))+geom_tile(color="black")+
scale_fill_manual(values=colorvec,na.value="white")plot
ggplot_mainplot1<-ggplot_mainplot1+theme_classic()+
theme(axis.text.x=element_text(size=5,face="bold",angle=45,vjust=0.8,hjust=0.8,family="Helvetica"),
axis.text.y=element_text(size=5,face="bold",vjust=0.8,family="Helvetica"),axis.title.y=element_blank(),
axis.line.y=element_blank(),axis.line.x=element_blank(),
axis.title.x=element_blank(),legend.key.size=unit(0.7,"cm"),
legend.title=element_blank(),
legend.text=element_text(size=5),
panel.background = element_rect(fill='transparent'),
plot.background = element_rect(fill='transparent', color=NA),
panel.grid.major = element_blank(), panel.grid.minor = element_blank(),
legend.background = element_rect(fill='transparent'),
legend.key.height= unit(0.3, 'cm'),
legend.key.width= unit(0.2, 'cm'))#
ggplot_ivwdir1<-ggplot_ivwdir1+theme_classic()+
theme(axis.text.x=element_blank(),
axis.text.y=element_blank(),axis.title.y=element_blank(),axis.ticks.y=element_blank(),
axis.ticks.x=element_blank(),
axis.line.y=element_blank(),axis.line.x=element_blank(),
axis.title.x=element_blank(),legend.key.size=unit(0.7,"cm"),
legend.text=element_text(size=5), legend.title=element_text(size=5),
panel.background = element_rect(fill='transparent'),
plot.background = element_rect(fill='transparent', color=NA),
legend.background = element_rect(fill='transparent'),
legend.key.height= unit(0.3, 'cm'),
legend.key.width= unit(0.2, 'cm'))#
ggplot_celltype1<-ggplot_celltype1+theme_classic()+
theme(axis.text.x=element_text(size=5,face="bold",angle=45,vjust=0.8,hjust=0.8,family="Helvetica"),
axis.text.y=element_blank(),axis.title.y=element_blank(),axis.ticks.y=element_blank(),
axis.line.y=element_blank(),axis.line.x=element_blank(),
axis.title.x=element_blank(),legend.key.size=unit(0.7,"cm"),
legend.title=element_blank(),
legend.text=element_text(size=5),
panel.background = element_rect(fill='transparent'),
plot.background = element_rect(fill='transparent', color=NA),
legend.background = element_rect(fill='transparent'),
legend.key.height= unit(0.3, 'cm'),
legend.key.width= unit(0.2, 'cm'))#
ggplot_mainplot1_legend<-get_legend(ggplot_mainplot1)
celltype_legend<-get_legend(ggplot_celltype1)
ivw_dir<-get_legend(ggplot_ivwdir1)
ggplot_ivwdir1<-ggplot_ivwdir1+theme(plot.margin = unit(c(0,0,0,0), "cm"),legend.position = "none")
ggplot_mainplot1<-ggplot_mainplot1+theme(plot.margin = unit(c(0,0,0,0), "cm"),legend.position = "none")
ggplot_celltype1<-ggplot_celltype1+theme(plot.margin = unit(c(0,0,0,0), "cm"),legend.position="none")
g<-plot_grid(ggplot_mainplot1,ggplot_celltype1,ggplot_ivwdir1, align = "h", ncol =3, rel_widths = c(0.008,0.005,0.001))
g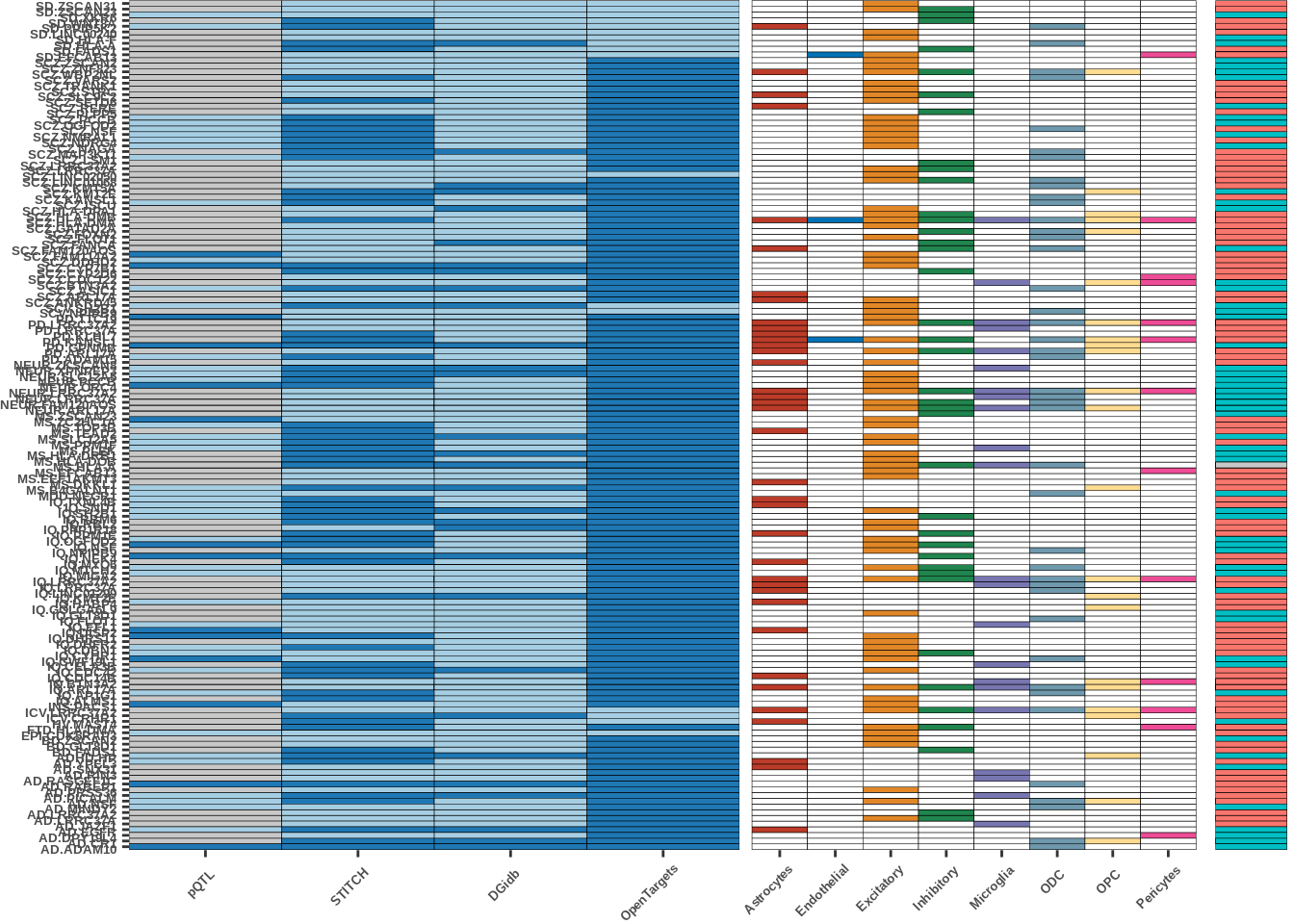
Figure 4b
suppressMessages(library(UpSetR))
library(dplyr)
suppressMessages(library(ComplexUpset))
library(tidyr)
color_pal=ggsci::pal_nejm("default")(8)
colorvec<-c(Astrocytes=color_pal[1],
Endothelial=color_pal[2],
Excitatory=color_pal[3],
Inhibitory=color_pal[4],Microglia=color_pal[5],
ODC=color_pal[6],OPC=color_pal[7],Pericytes=color_pal[8])
full<-read.table("data/COLOC_MR_RESULTS//2022-10-25_FULL_MR_RES.txt")
gwas<-unique(full$GWAS)
genes<-unique(full$gene)
resdf<-data.frame(gene=genes)
for(i in 1:length(gwas)){
tmp_genes<-full[full$GWAS==gwas[i],]$gene
resdf<-cbind(resdf,genes %in% tmp_genes)
}
rownames(resdf)<-resdf$gene
resdf$gene<-NULL
colnames(resdf)<-gwas
plot<-upset(resdf,gwas,width_ratio=0.1,height_ratio = 0.4,set_size=FALSE,
themes=upset_default_themes(text=element_text(size=6,family="Helvetica")),
base_annotations=list(
'Intersection size'=intersection_size(color="#000000",size=0.25,
mapping=aes(fill="bars_color"),
text=list(size=5/(14/5),family="Helvetica"))
+scale_fill_manual(values=c("bars_color"="#1F78B4"),guide="none")
+theme(plot.background=element_blank(),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank())),
stripes="white",matrix=(
intersection_matrix(
geom=geom_point(size=0.5),
segment=geom_segment(size=0.3)
))
)
plot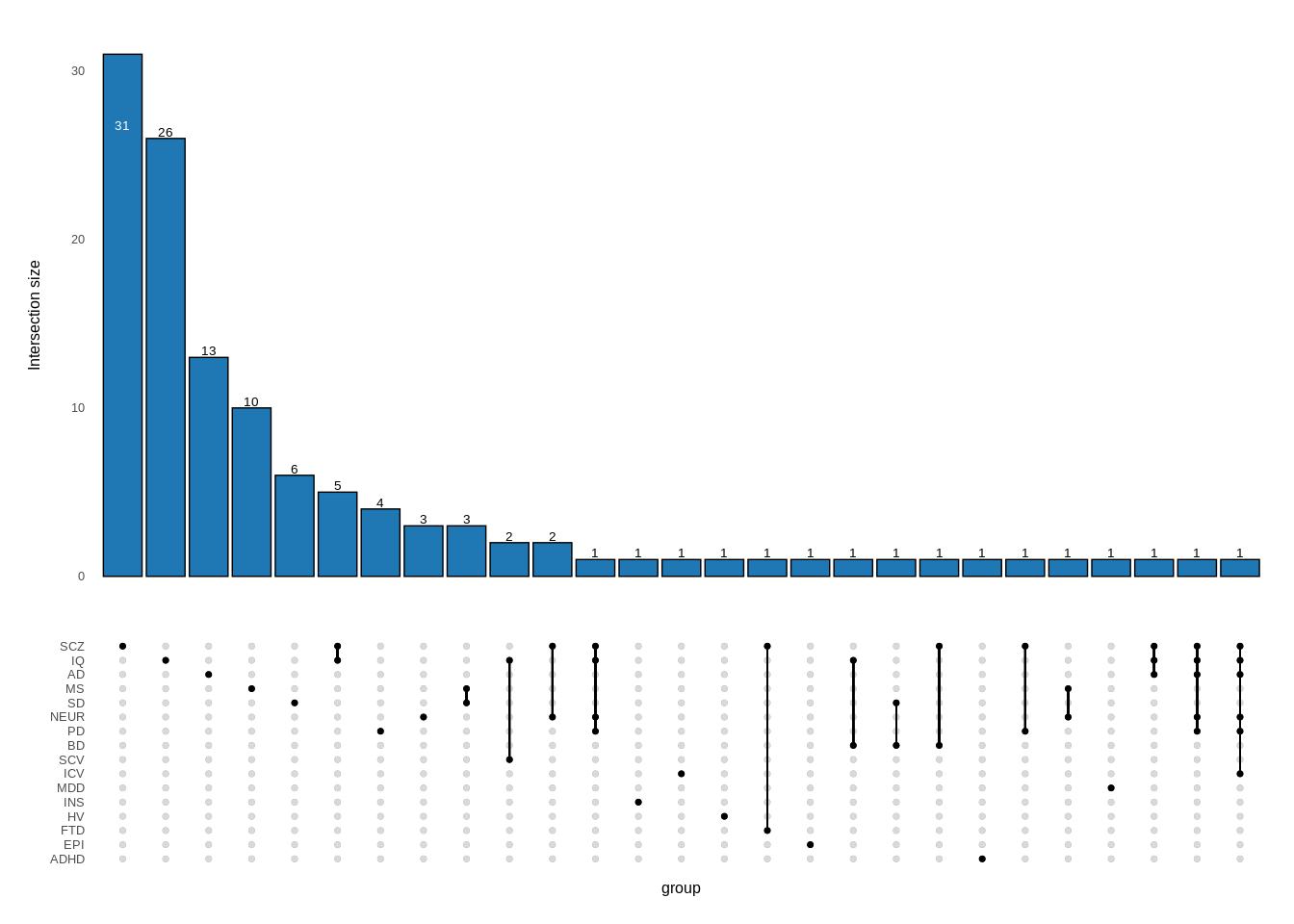
SUPPLEMENTARY FIGURES
Suppl. Fig. 1
Suppl. Fig. 1a
metadata<-readRDS("data/METADATA/Final_Seurat_129samples_15May2022_metadata.rds")
##this sample was excluded earlier
metadata<-metadata[!metadata$Sample_ID %in% "O141",]
samples<-unique(metadata$Sample_ID)
df<-data.frame(samples=samples,new_id=1:length(samples))
metadata$new_id<-paste0("Sample ",as.character(df[match(metadata$Sample_ID,df$samples),]$new_id))
metadata$new_id<-factor(metadata$new_id, levels =unique(metadata$new_id))
tmp<-metadata
color_panel<-ggsci::pal_nejm("default")(8)
p<-ggplot(tmp,aes(x=new_id,fill=CellType))+geom_bar(color="black")+
scale_fill_manual(values=color_panel)+
scale_y_continuous(expand=c(0,0))+
theme_classic()+theme(axis.text.x=element_text(size=5,face="bold",angle=90),
plot.title = element_text(size=5,face="bold"),
axis.text.y=element_text(size=5,face="bold"),
axis.title.y=element_blank(),
axis.title.x=element_blank(),
legend.key.size=unit(0.7,"cm"),
legend.position="none",
legend.text=element_text(size=15),panel.background = element_rect(fill='transparent'), #transparent panel bg
plot.background = element_rect(fill='transparent', color=NA), #transparent plot bg
panel.grid.major = element_blank(), #remove major gridlines
panel.grid.minor = element_blank(), #remove minor gridlines
legend.background = element_rect(fill='transparent'), #transparent legend bg
legend.box.background = element_rect(fill='transparent'))+labs(title="Cell type distribution per indiviudal")
p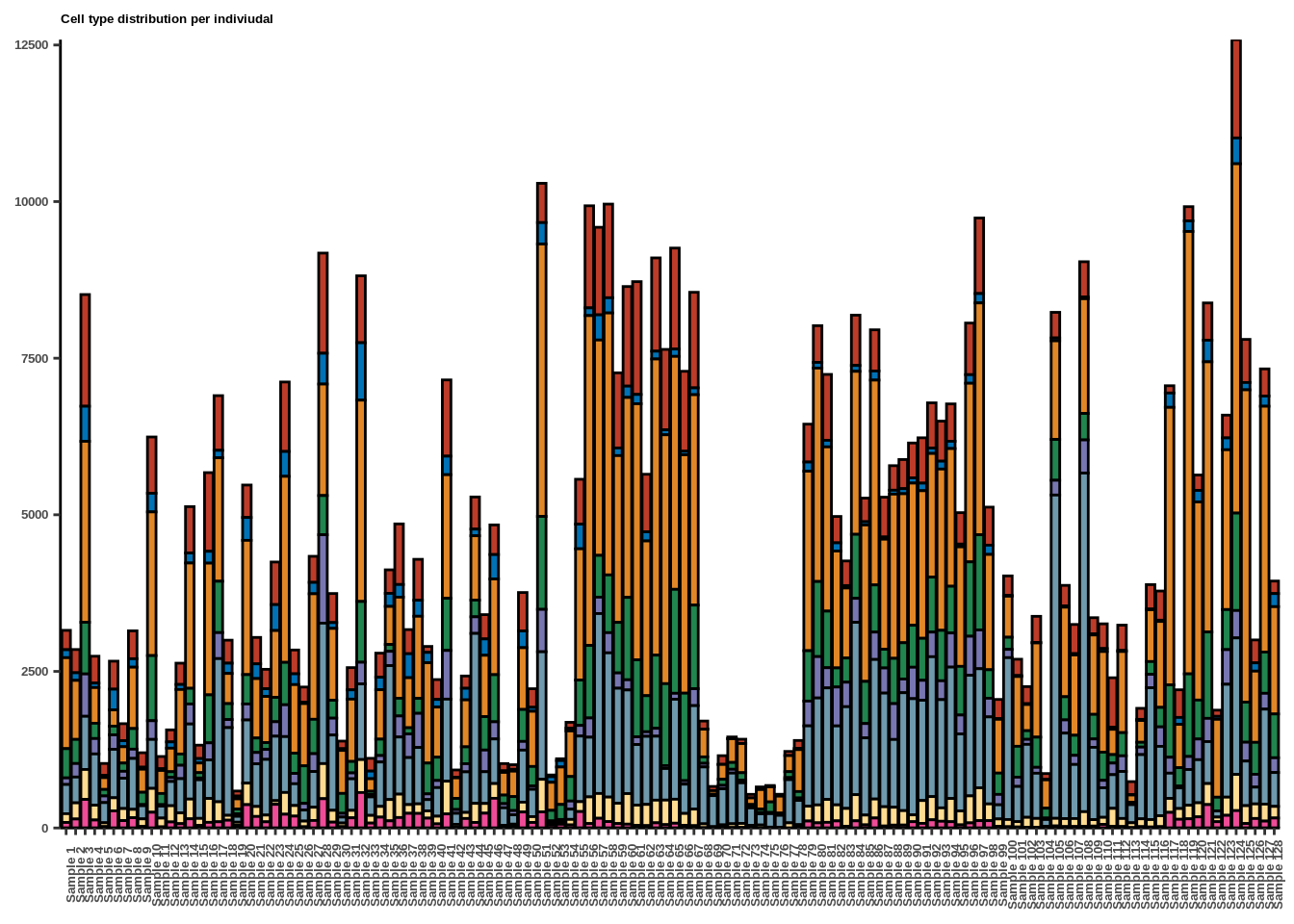
Suppl. Fig. 1b
library(ggrepel)
metadata<-readRDS("data/METADATA/Final_Seurat_129samples_15May2022_metadata.rds")
metadata<-metadata[!metadata$Sample_ID %in% "O141",]
df<-as.data.frame(table(metadata$CellType))
df$celltype<-df$Var1
library(dplyr)
df2 <- df %>%
mutate(csum = rev(cumsum(rev(Freq))),
pos =Freq/2 + lead(csum, 1),
pos = if_else(is.na(pos), Freq/2, pos))
p<-ggplot(df, aes(x="",y=Freq,fill=celltype)) +
geom_bar(stat="identity",colour="black",width=1)+
geom_label_repel(data = df2,
aes(y = pos, label = Freq),
size = 5/(14/5), nudge_x = 1, show.legend = FALSE)+
theme_classic()+theme(axis.text.x=element_blank(),
axis.line=element_blank(),
plot.title = element_text(size=7,face="bold"),
axis.text.y=element_blank(),
axis.title.y=element_blank(),
axis.title.x=element_blank(),
legend.key.size=unit(0.7,"cm"),
legend.position="none",
legend.text=element_text(size=5),panel.background = element_rect(fill='transparent'), #transparent panel bg
plot.background = element_rect(fill='transparent', color=NA), #transparent plot bg
panel.grid.major = element_blank(), #remove major gridlines
panel.grid.minor = element_blank(), #remove minor gridlines
legend.background = element_rect(fill='transparent'), #transparent legend bg
legend.box.background = element_rect(fill='transparent'))+labs(title="Total number of cells per cell type")+
ggsci::scale_fill_nejm()+ coord_polar("y", start=0)
p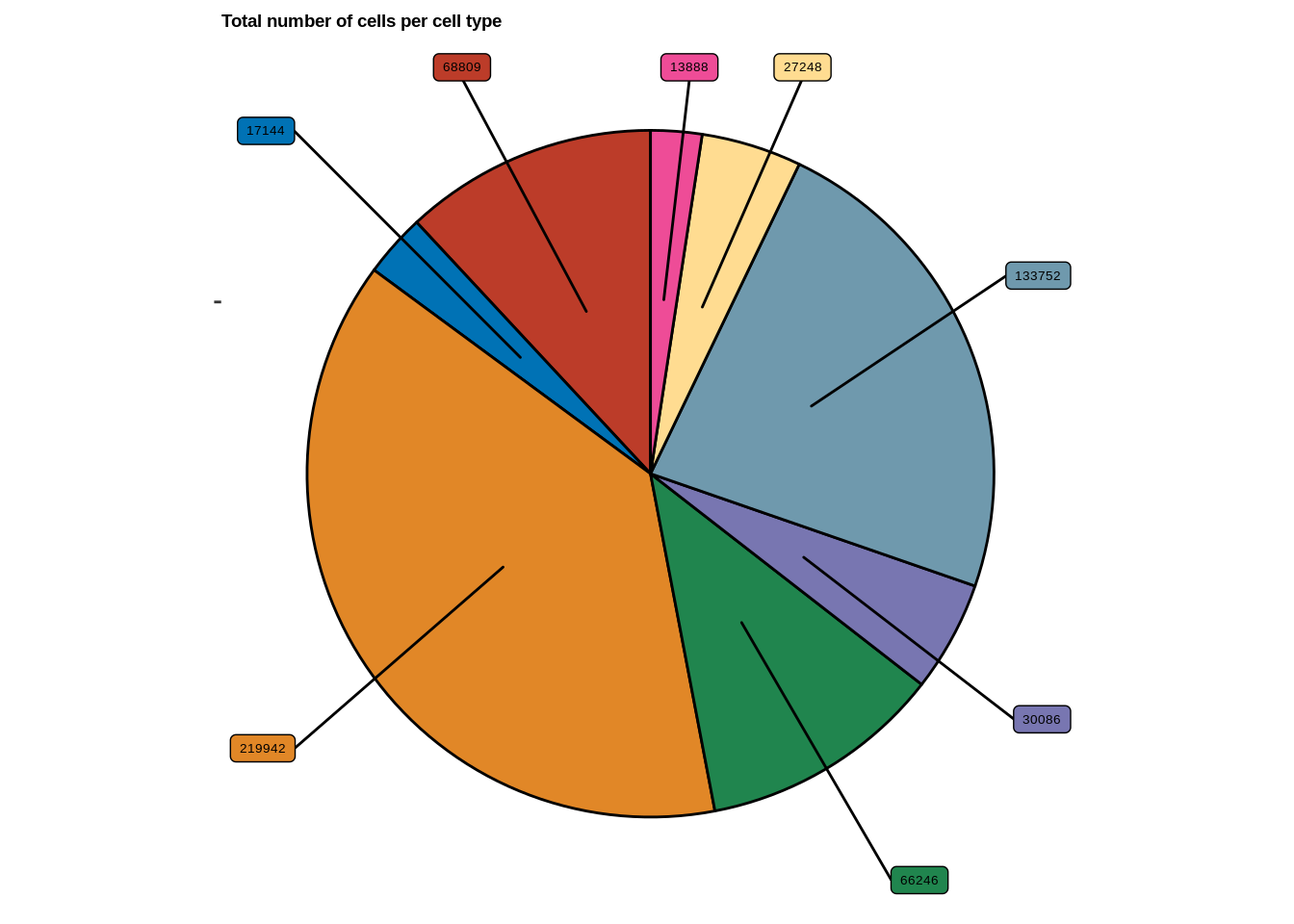
Suppl. Fig. 2
res<-read.table("data/EXT_DATASETS/METABRAIN//metabrain_replication.txt")
res$celltype<-c("Astrocytes","Endothelial","Excitatory","Inhibitory","Microglia","Oligo","OPC","Pericytes")
color_plane_1=ggsci::pal_nejm("default")(8)
colnames(res)<-c("total","replicated","percentage","totalinboth","celltype")
g<-ggplot()+
geom_point(res,mapping=aes(x=total,y=percentage,fill=celltype),size=2,shape=21)+
scale_fill_manual(values=c("Astrocytes"=color_plane_1[1],
"Endothelial"=color_plane_1[2],
"Excitatory"=color_plane_1[3],
"Inhibitory"=color_plane_1[4],
"Microglia"=color_plane_1[5],
"Oligo"=color_plane_1[6],
"OPC"=color_plane_1[7],
"Pericytes"=color_plane_1[8]))+
ylab("Percentage replication in metabrain")+
xlab("Total SNP gene pairs at 5% FDR\nalso assessed in metabrain")+
scale_y_continuous(breaks = scales::pretty_breaks(n = 10),limits=c(0,1),labels = function(x) paste0(x*100, "%"))+
scale_x_continuous(breaks = scales::pretty_breaks(n = 7))+
theme_classic()+theme(axis.text.x=element_text(size=5,family="Helvetica",angle=45,vjust=0.6,face="bold"),
axis.text.y=element_text(size=5,family="Helvetica",face="bold"),
axis.title.y=element_text(size=6,family="Helvetica",margin=margin(r=20),face="bold"),
axis.title.x=element_text(size=6,family="Helvetica",margin=margin(t=20),face="bold"),
legend.key.size=unit(0.25,"cm"),
# legend.position="none",
legend.text=element_text(size=5),legend.title=element_blank())
g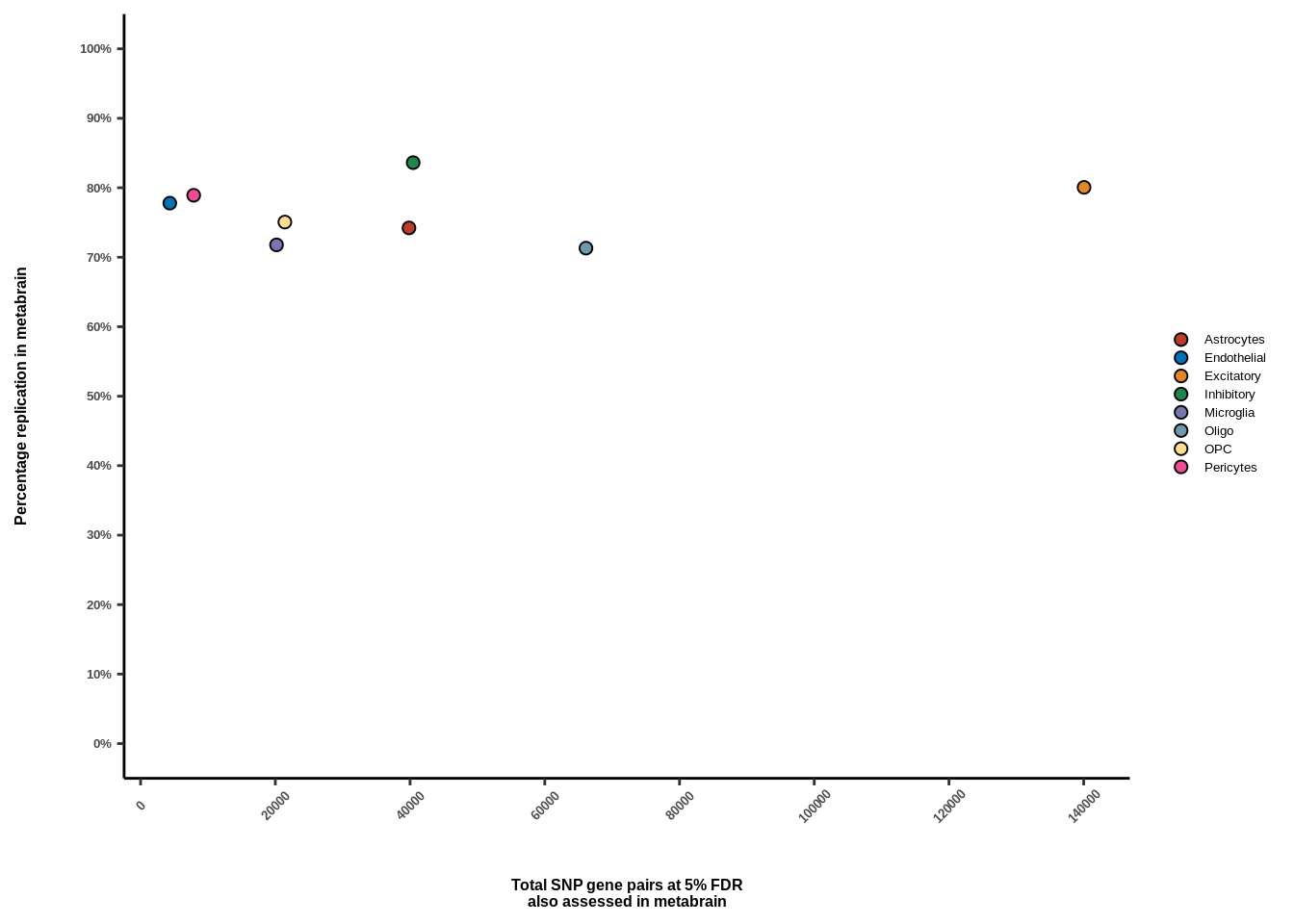
Suppl. Fig. 3
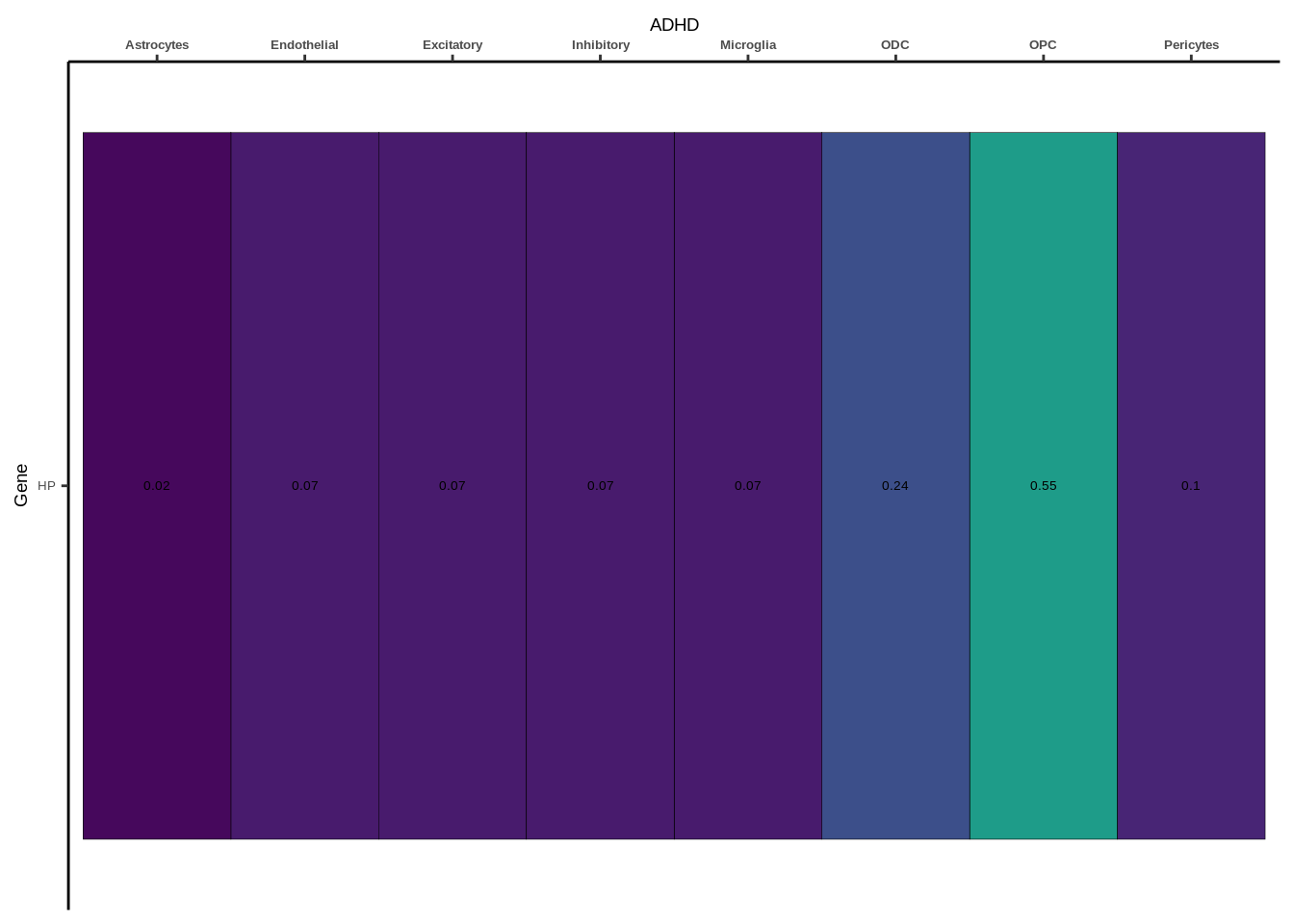
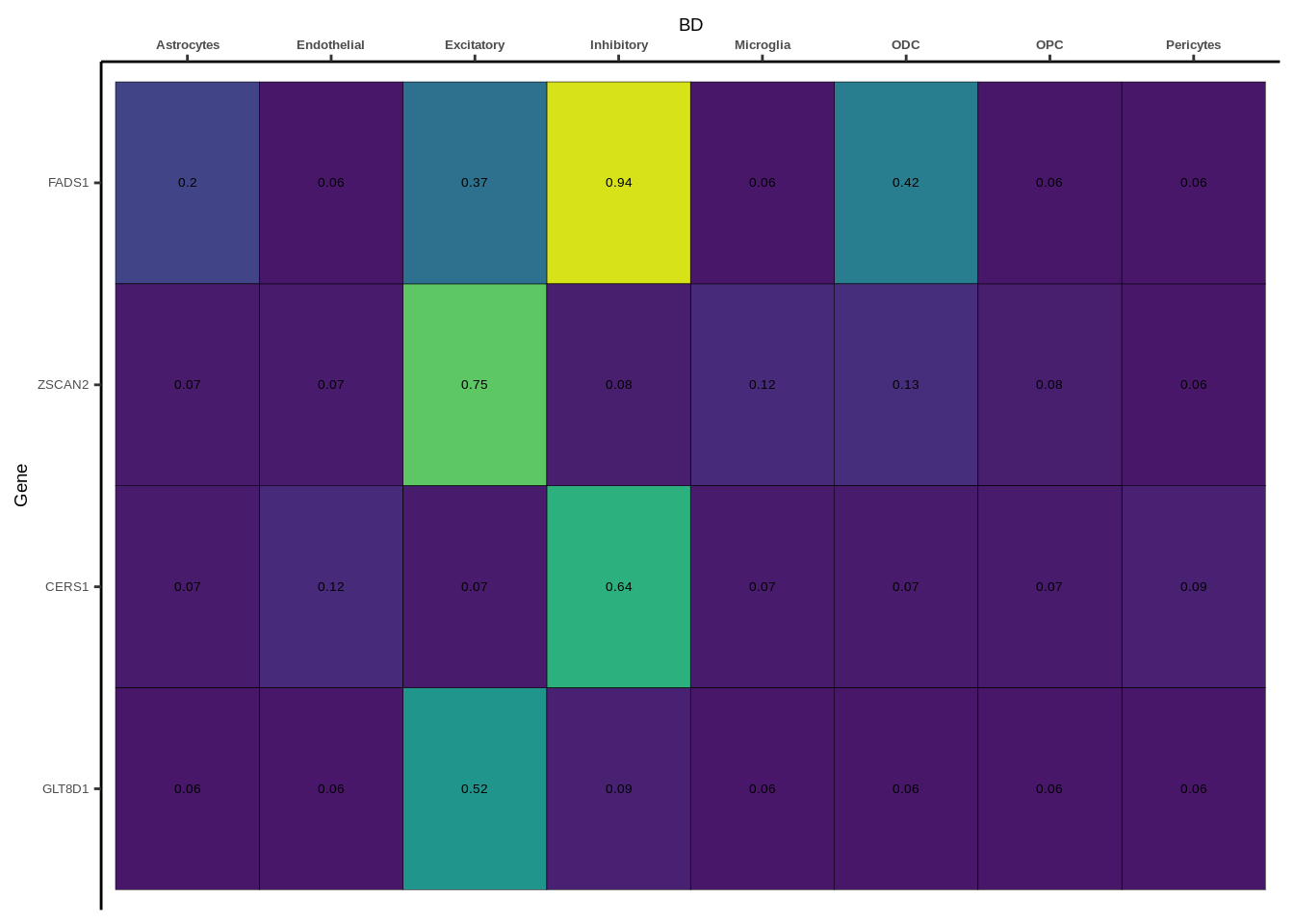
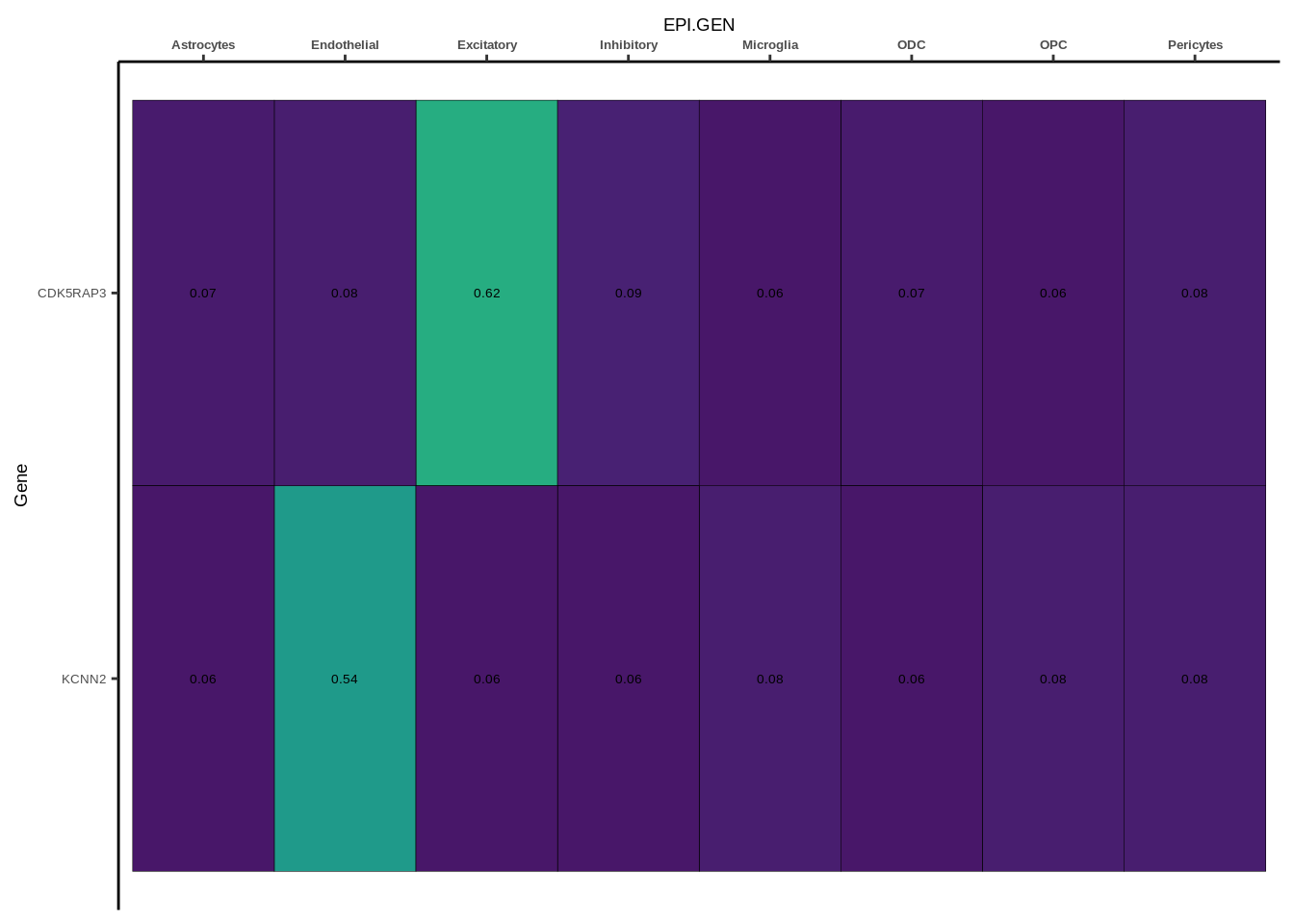
coloc_df<-read.table("data/COLOC_MR_RESULTS//2022-10-25_FULL_COLOC_RES.txt")
coloc_list<-split(coloc_df,coloc_df$GWAS)
heatmap_list<-list()
size_vector<-vector()
for(i in 1:length(coloc_list)){
x<-coloc_list[[i]]
if(length(x[x$PP.H4.abf>0.5,]$gene)==0){
heatmap_list[[i]]<-0
size_vector<-c(size_vector,0)
}else{
genes<-x[x$PP.H4.abf>0.5,]$gene
x<-x[x$gene %in% genes,]
x$Gene<-factor(x$gene,levels=unique(x$gene))
g<-ggplot(x,aes(x=celltype,y=Gene,fill=PP.H4.abf))
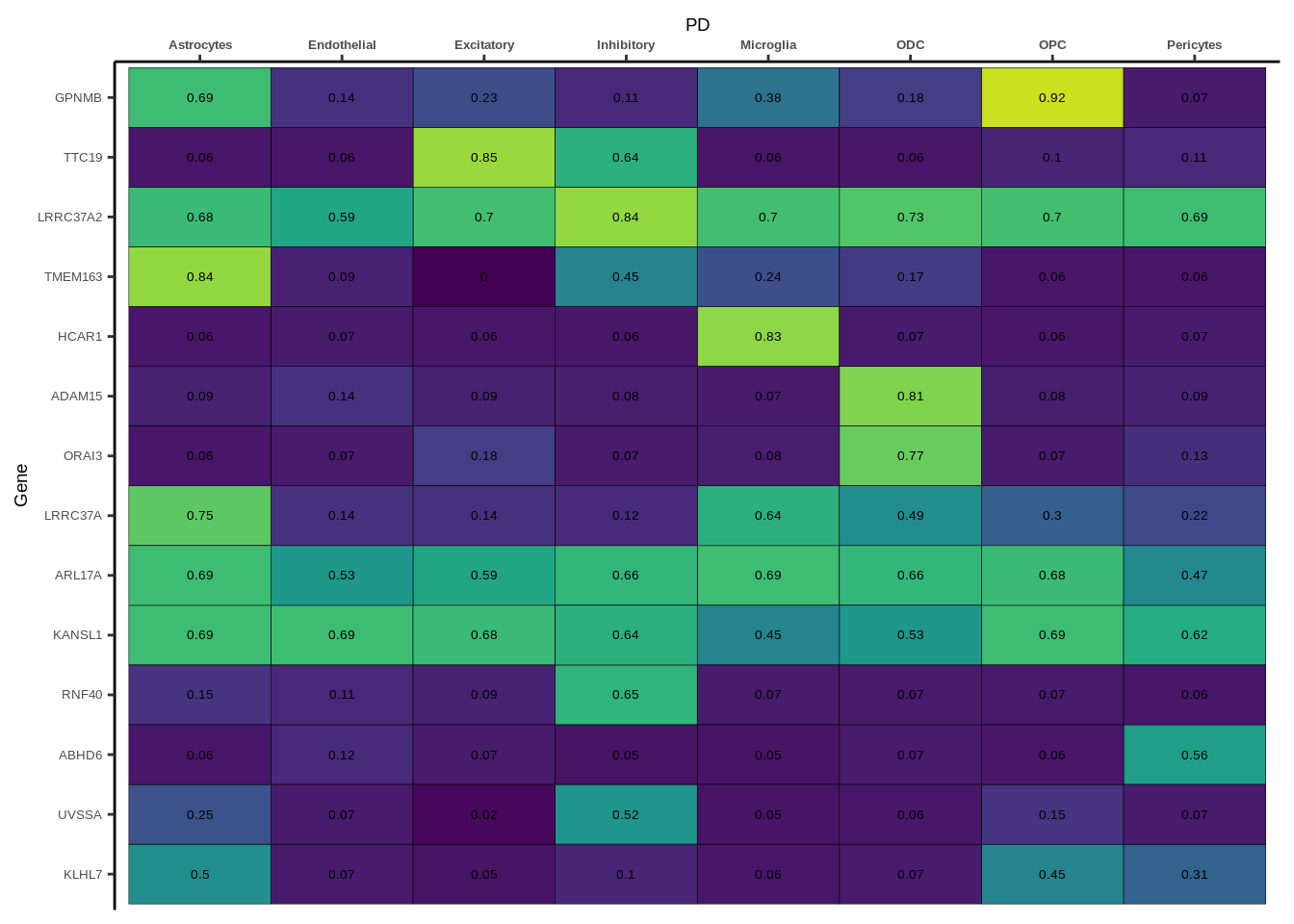
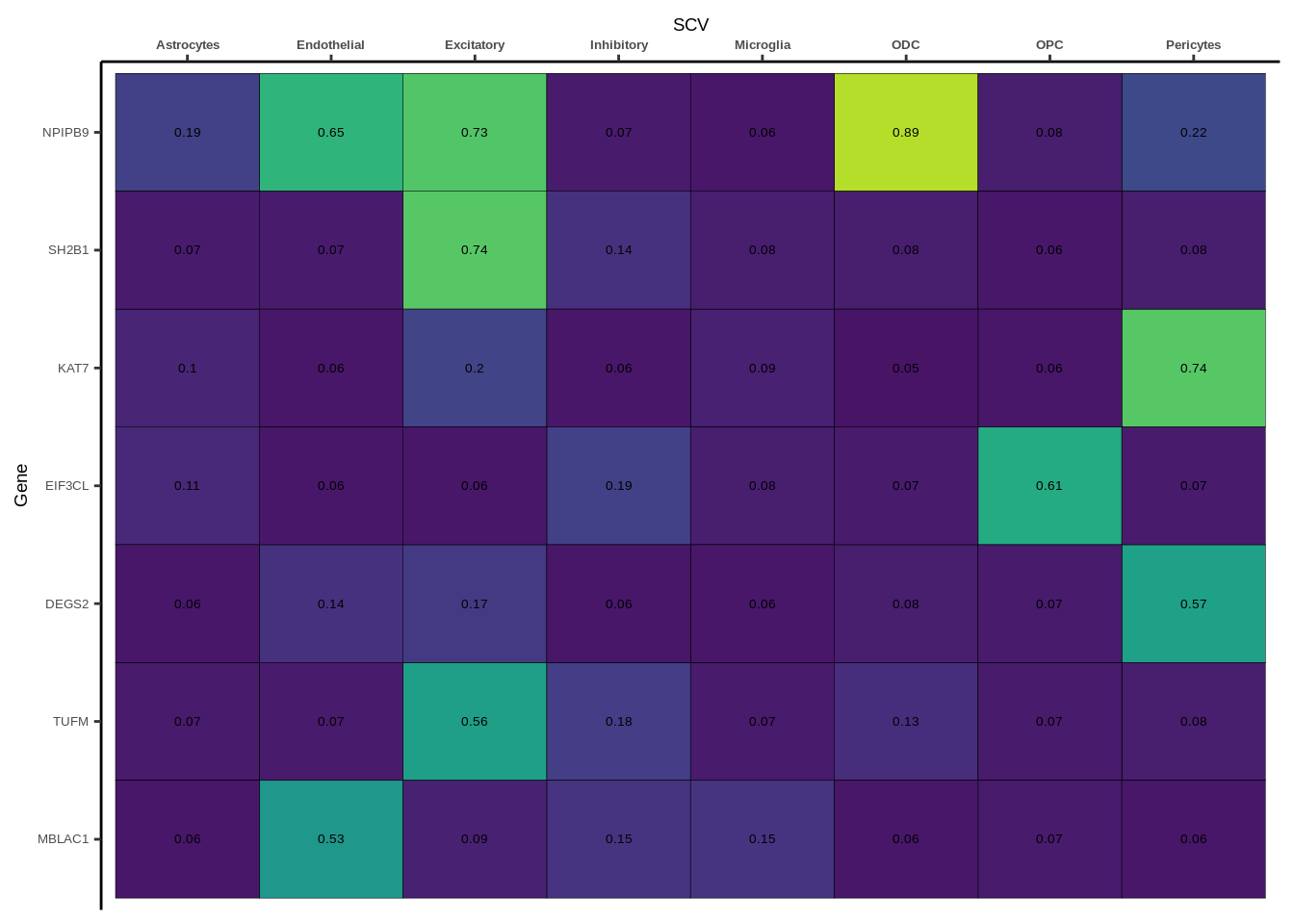
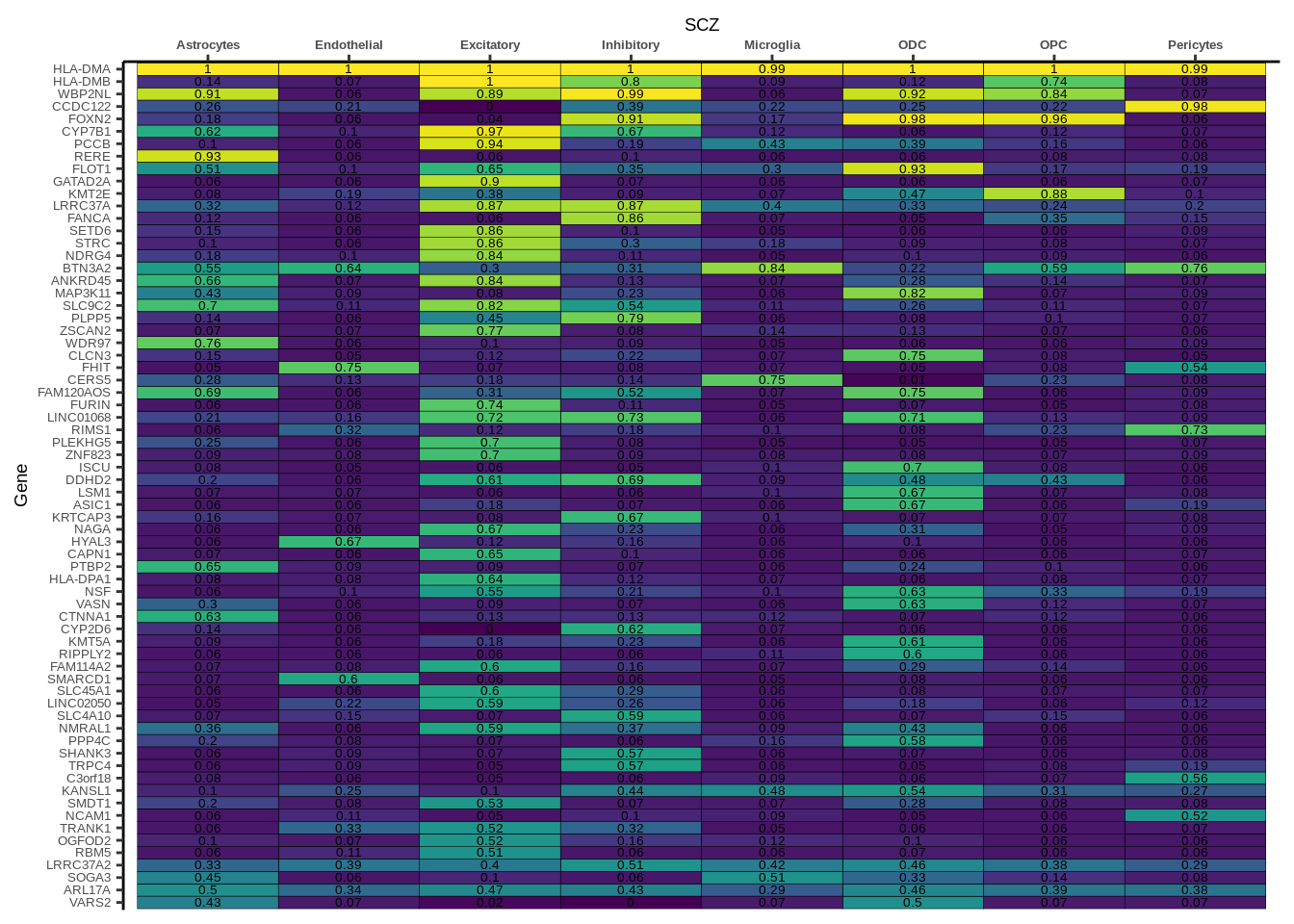
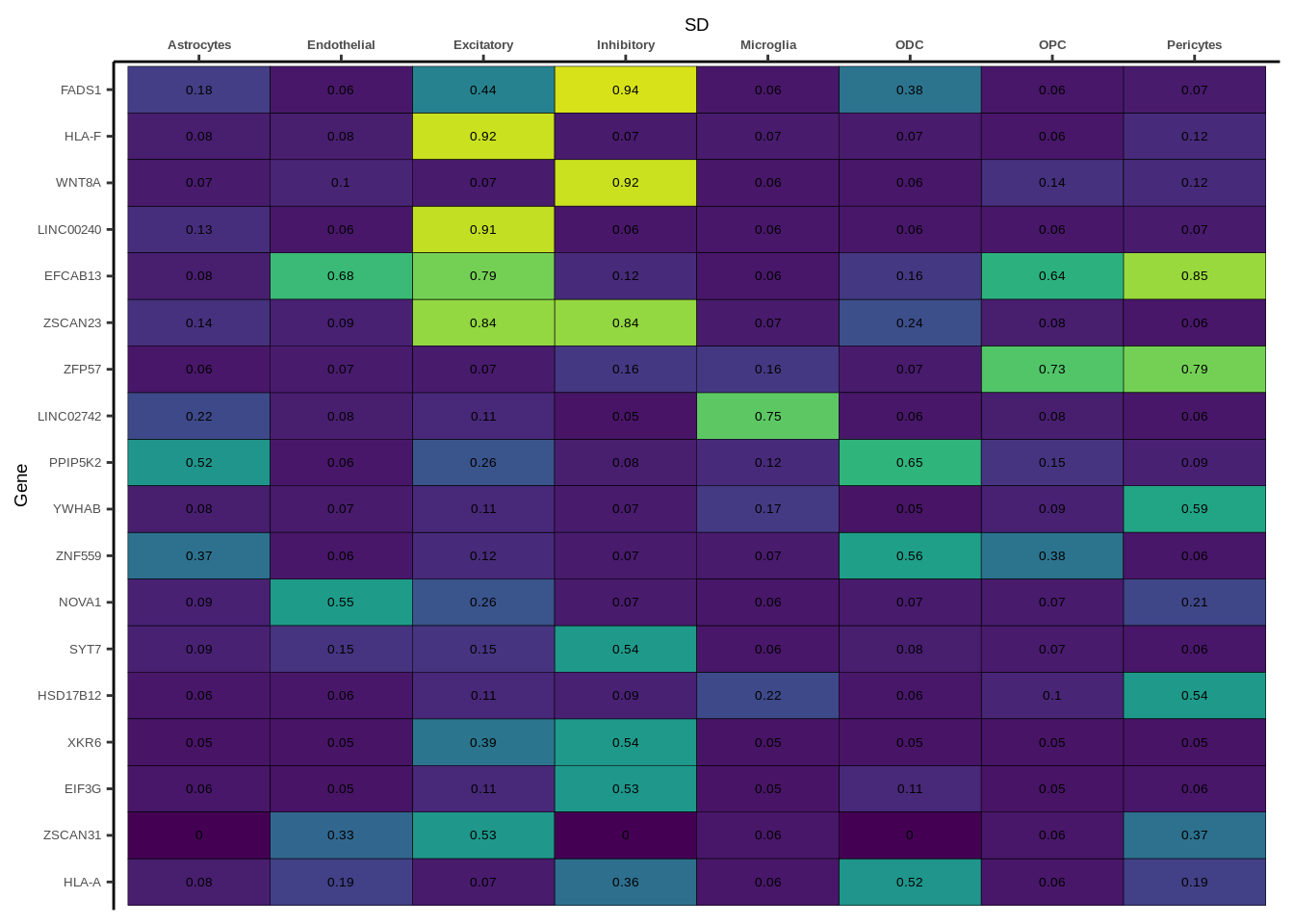
h1<-g+geom_tile(aes(fill=round(PP.H4.abf,2)),colour="black")+geom_text(aes(label = round(PP.H4.abf, 2)),size=5*0.36,family="Helvetica")+
scale_fill_viridis(limits=c(0,1))+
theme_classic()+scale_y_discrete(limits=rev)+scale_x_discrete(position="top")+xlab(x$GWAS[1])+
theme(axis.text.x=element_text(size=5,family="Helvetica",face="bold"),
axis.text.y=element_text(size=5,family="Helvetica"),
axis.title=element_text(size=7,family="Helvetica"),
legend.position="none")
heatmap_list[[i]]<-h1
size_vector<-c(size_vector,length(unique(genes)))
}
}
names(heatmap_list)<-unique(coloc_df$GWAS)
options(warn=-1)
for(i in 1:length(heatmap_list)){
if(class(heatmap_list[[i]])!="numeric"){
print(heatmap_list[[i]])
}
}
| Version | Author | Date |
|---|---|---|
| 7389e87 | Alexander Haglund | 2022-12-01 |
| bb50a67 | Alexander Haglund | 2022-12-01 |
| 10dd725 | Alexander Haglund | 2022-12-01 |
| 3c46f4b | Alexander Haglund | 2022-12-01 |
| fd90855 | Alexander Haglund | 2022-12-01 |
| 016f567 | Alexander Haglund | 2022-12-01 |
| 43e9b1f | Alexander Haglund | 2022-12-01 |
| 3c08408 | Alexander Haglund | 2022-12-01 |
| 4fcf3ea | Alexander Haglund | 2022-12-01 |
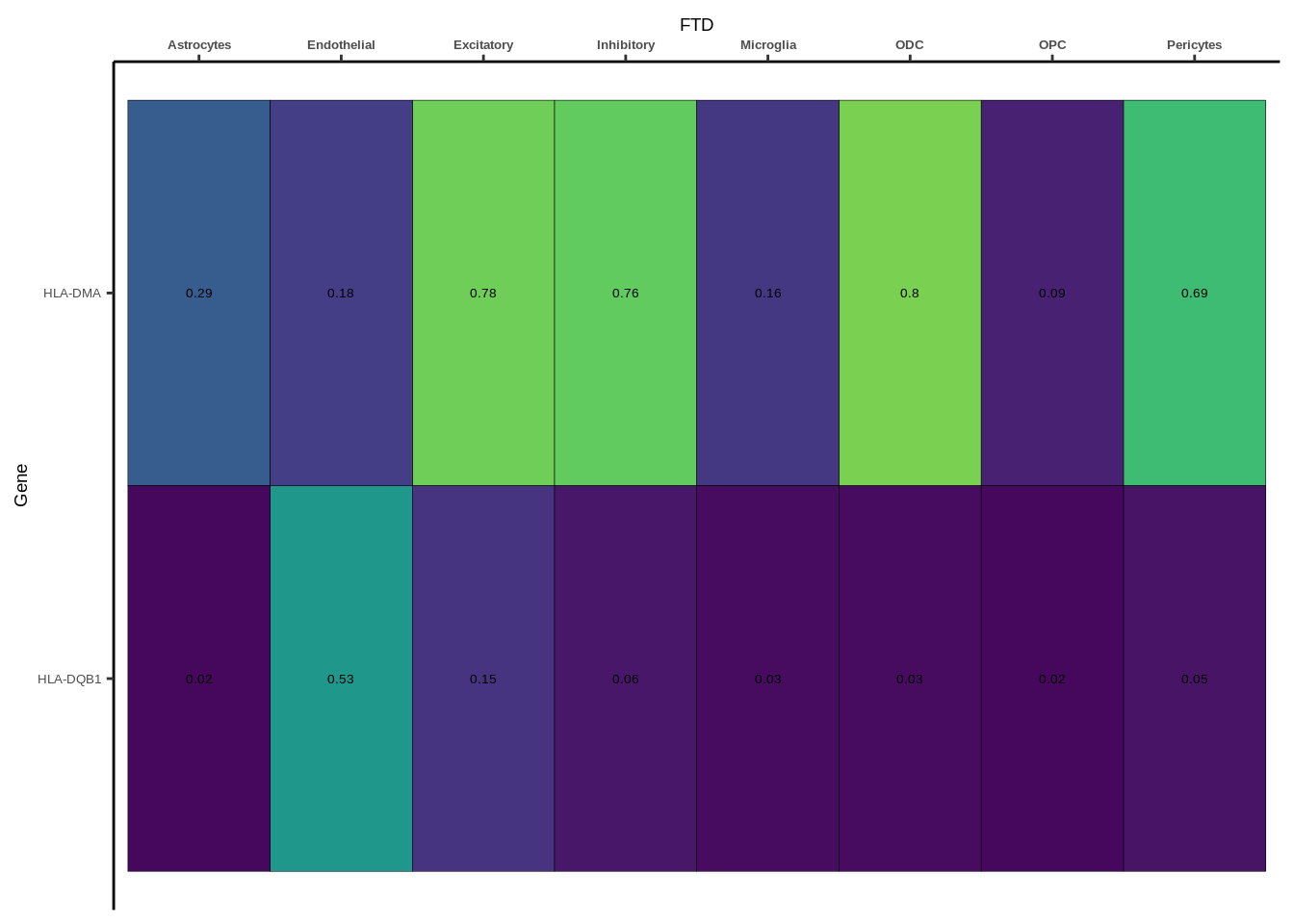
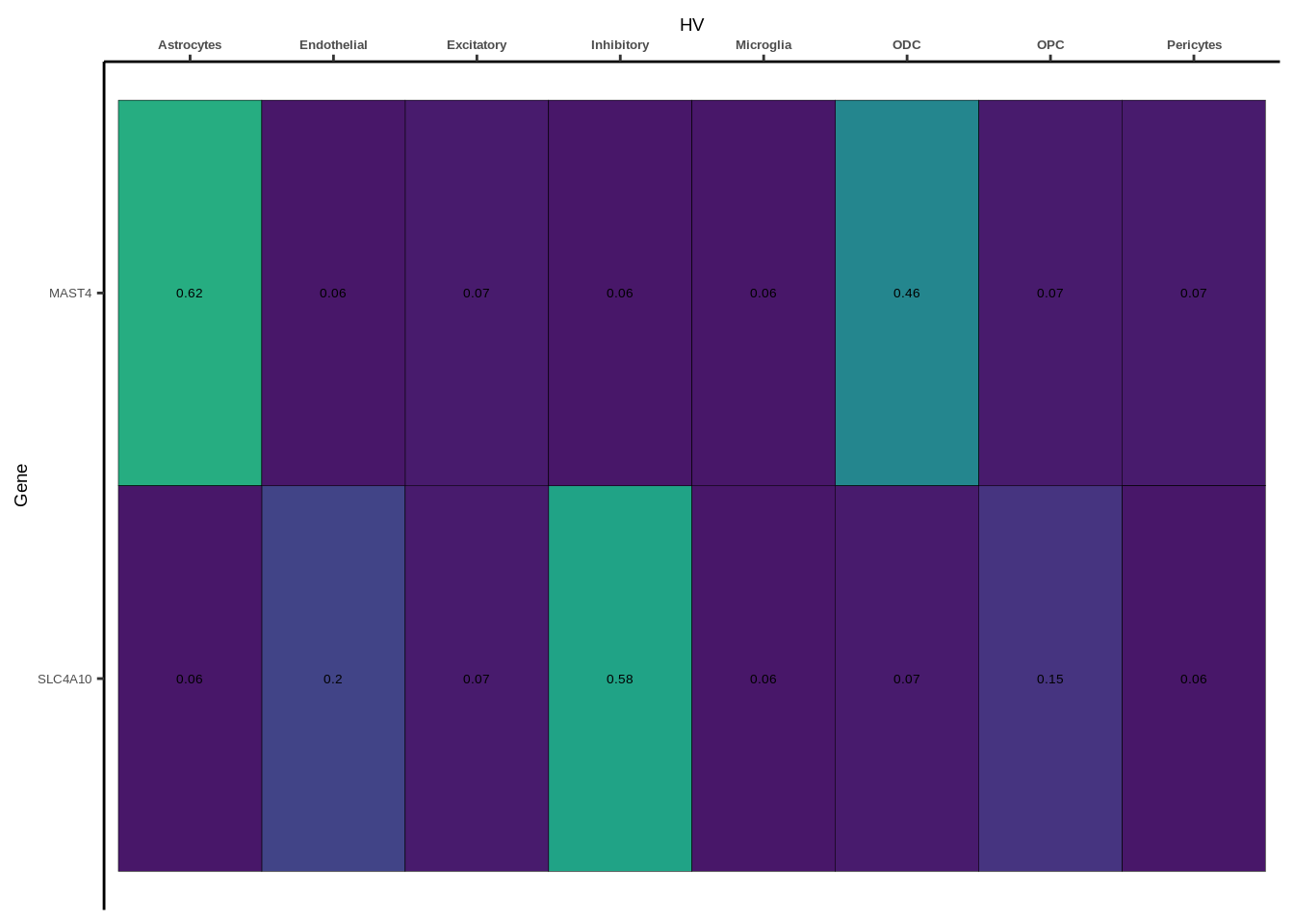
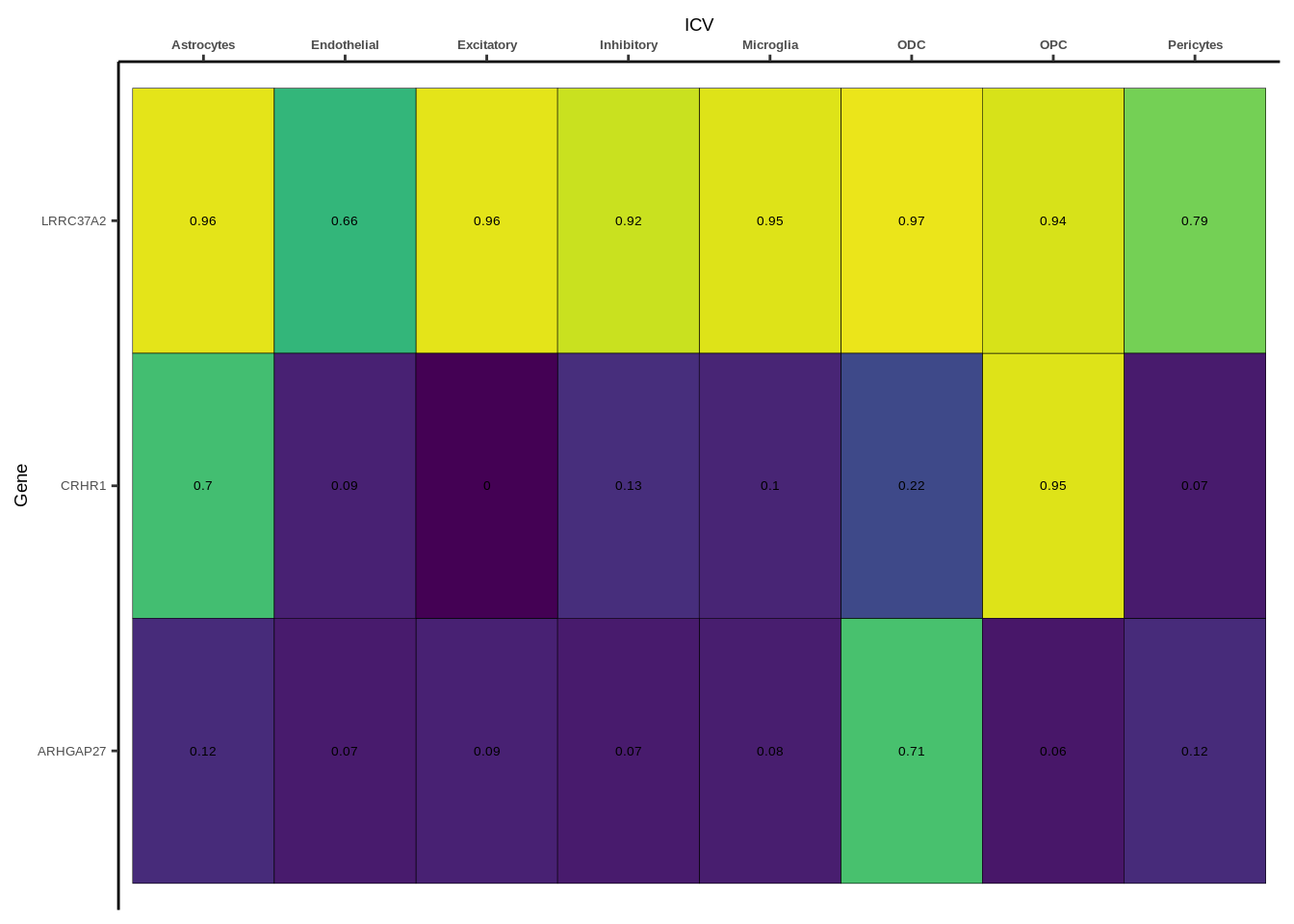
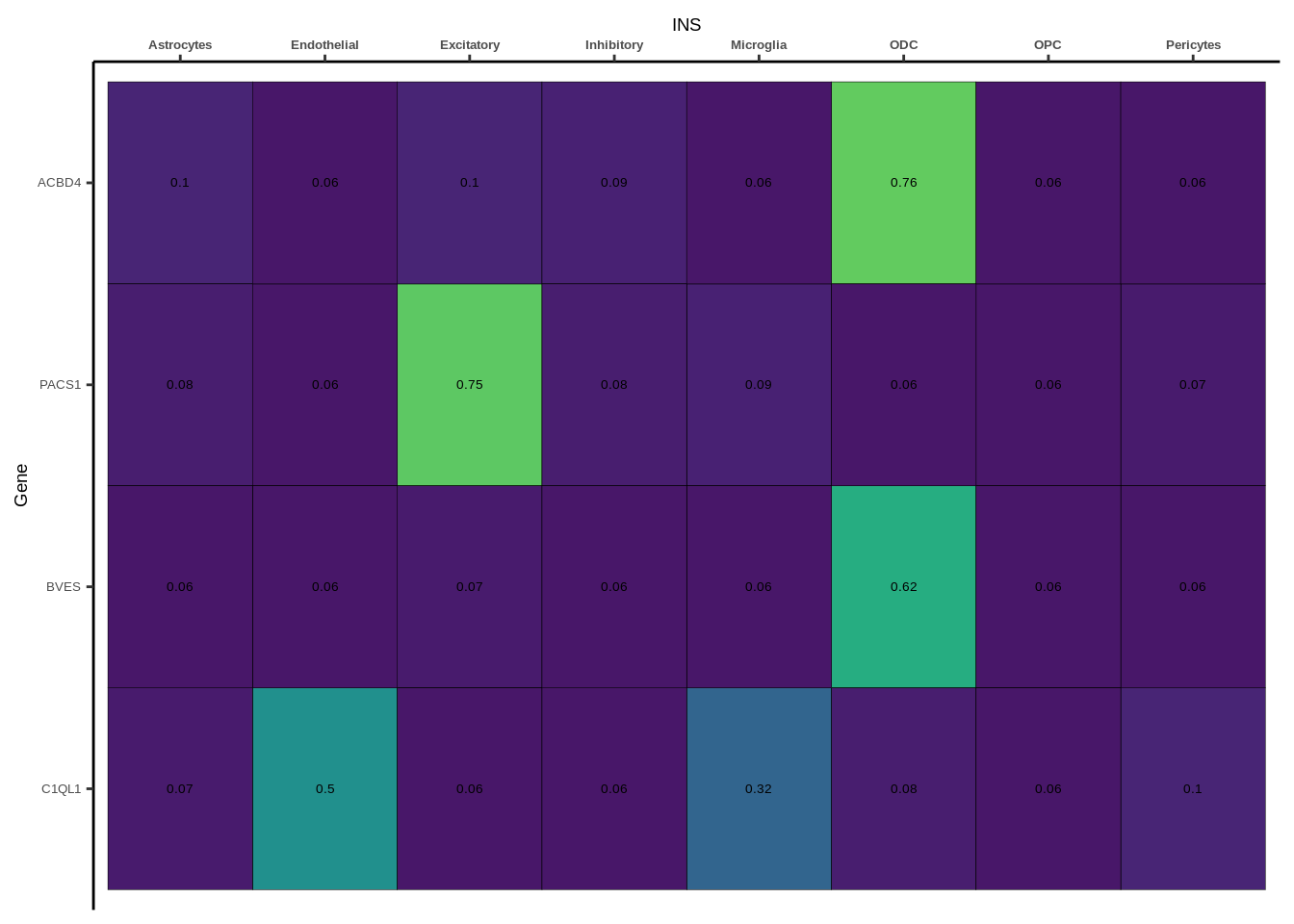
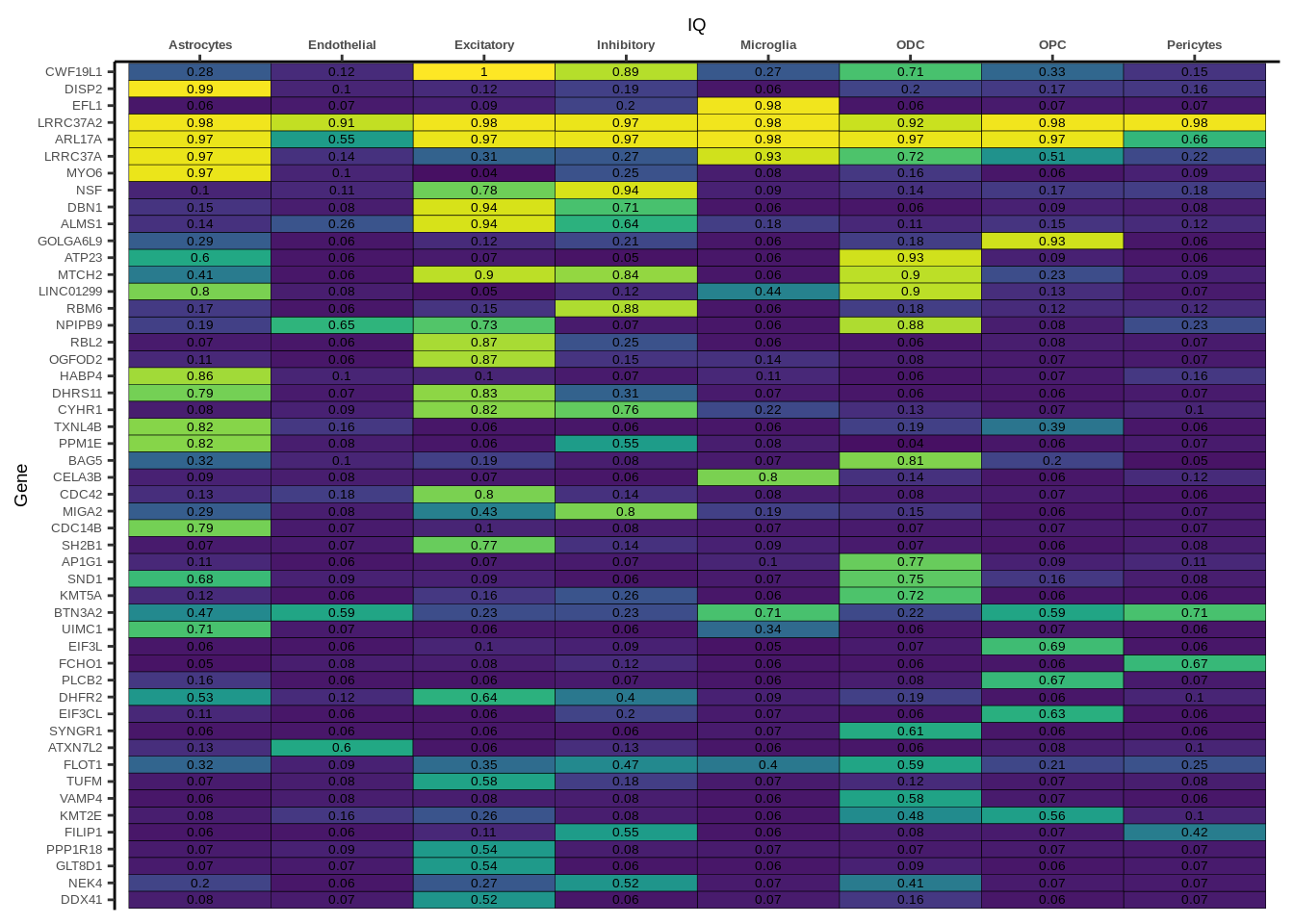
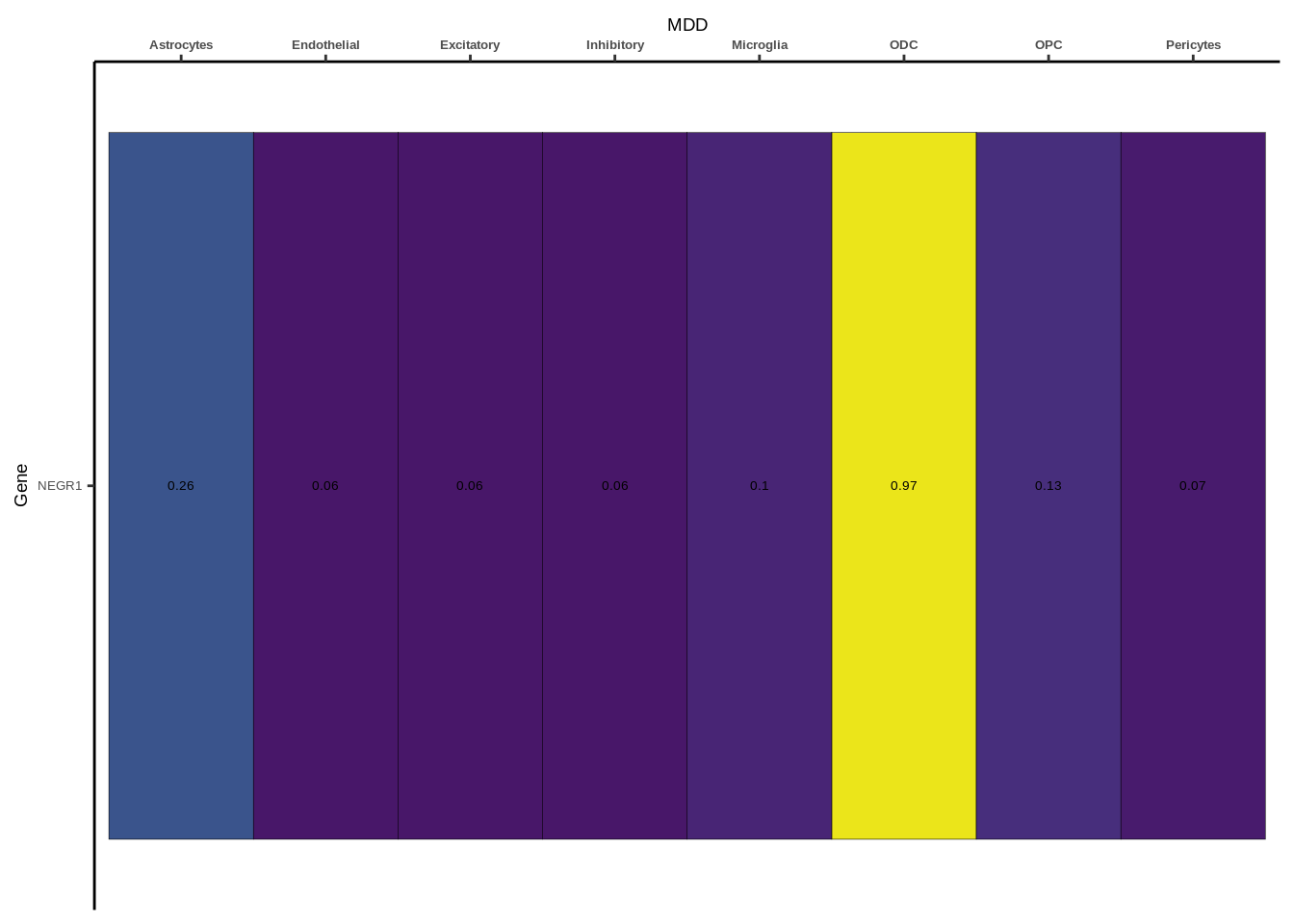
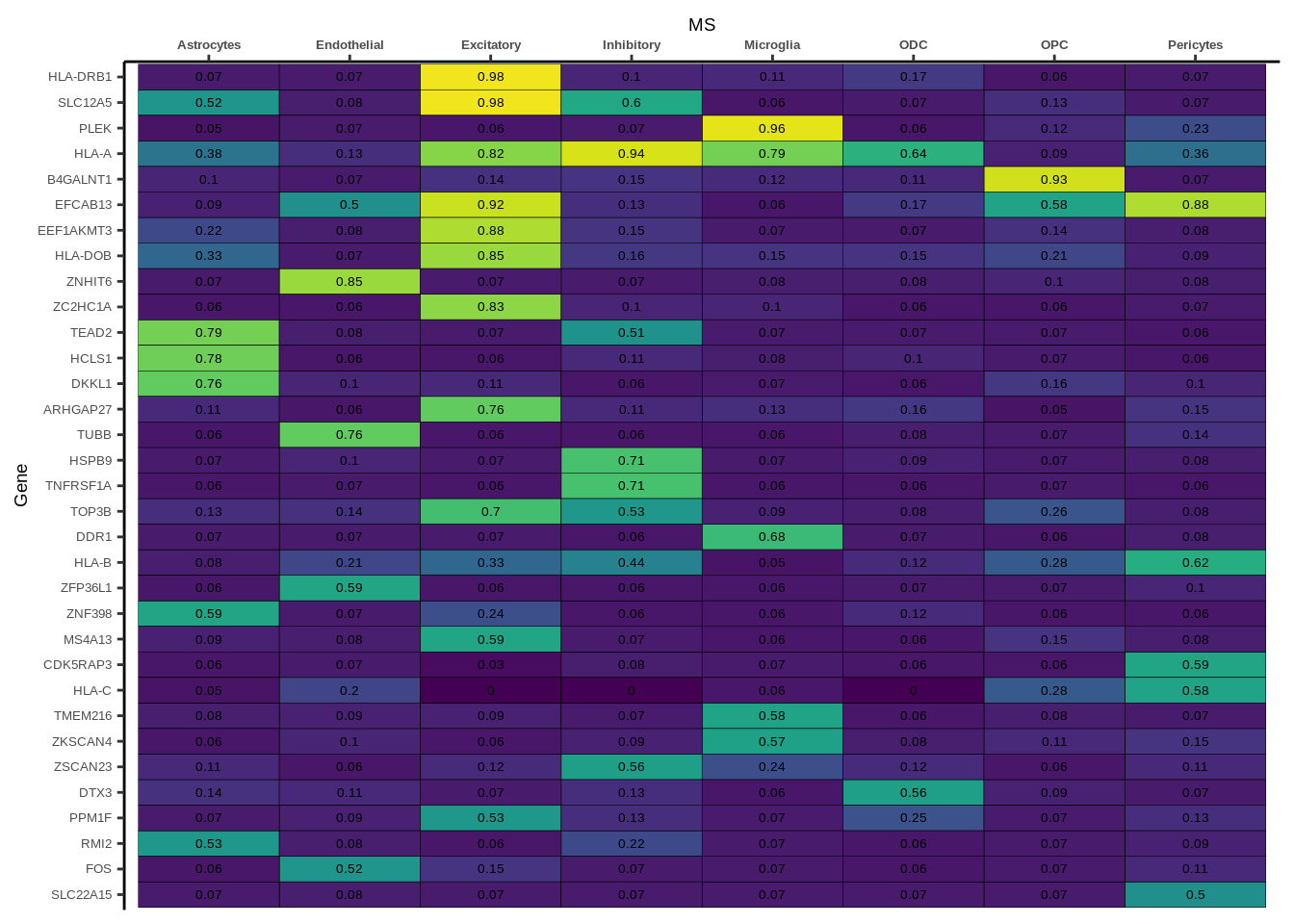
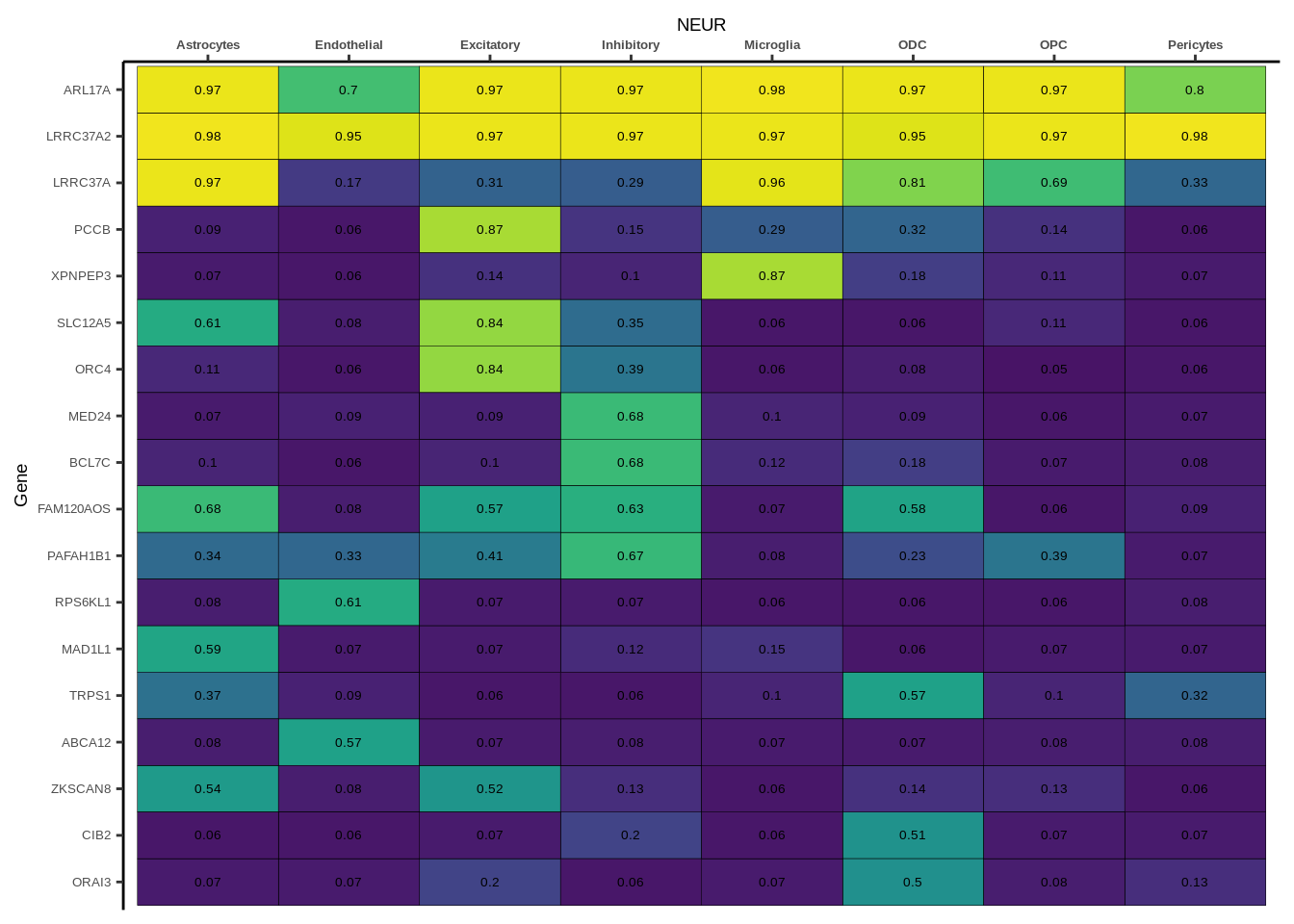
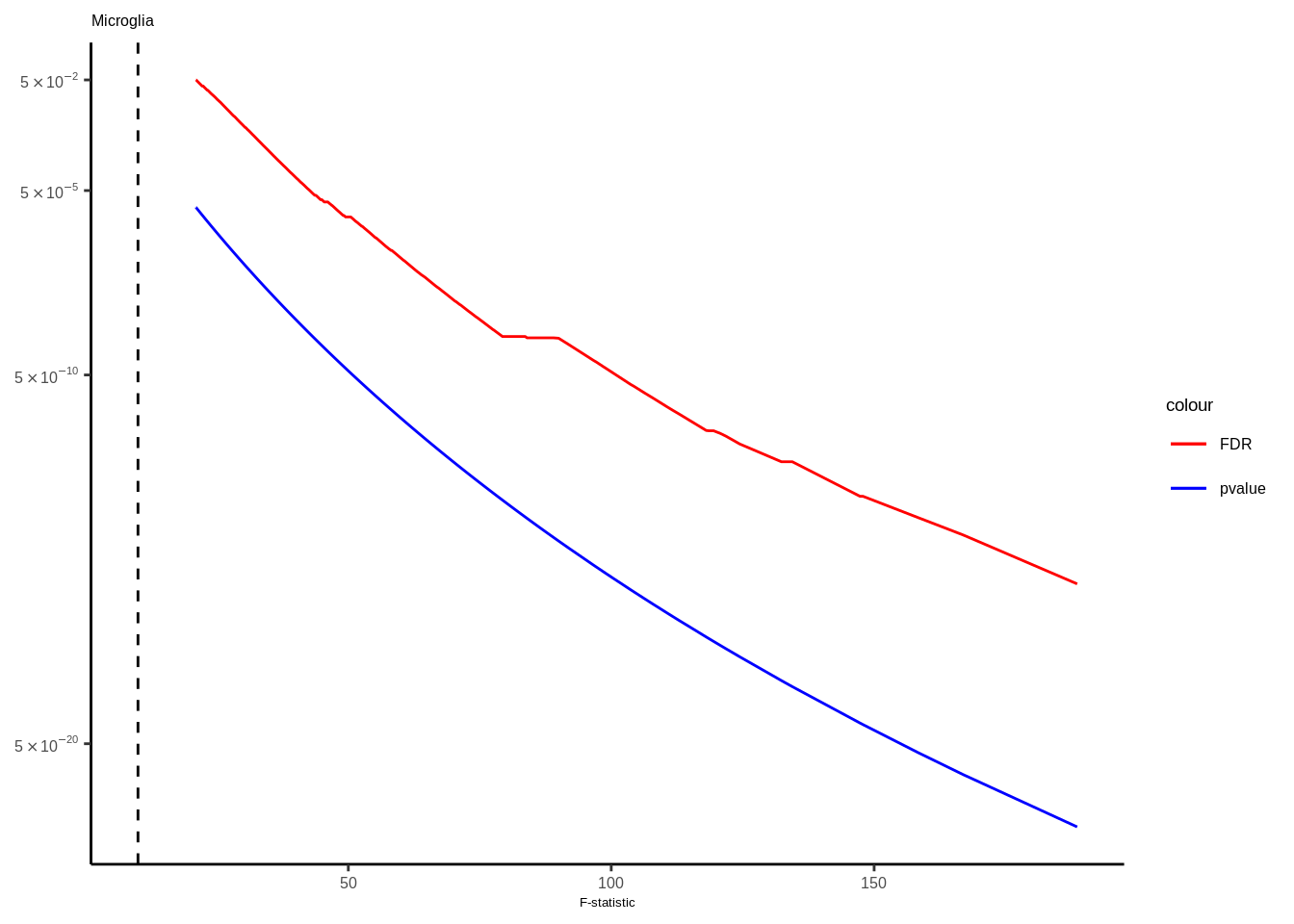
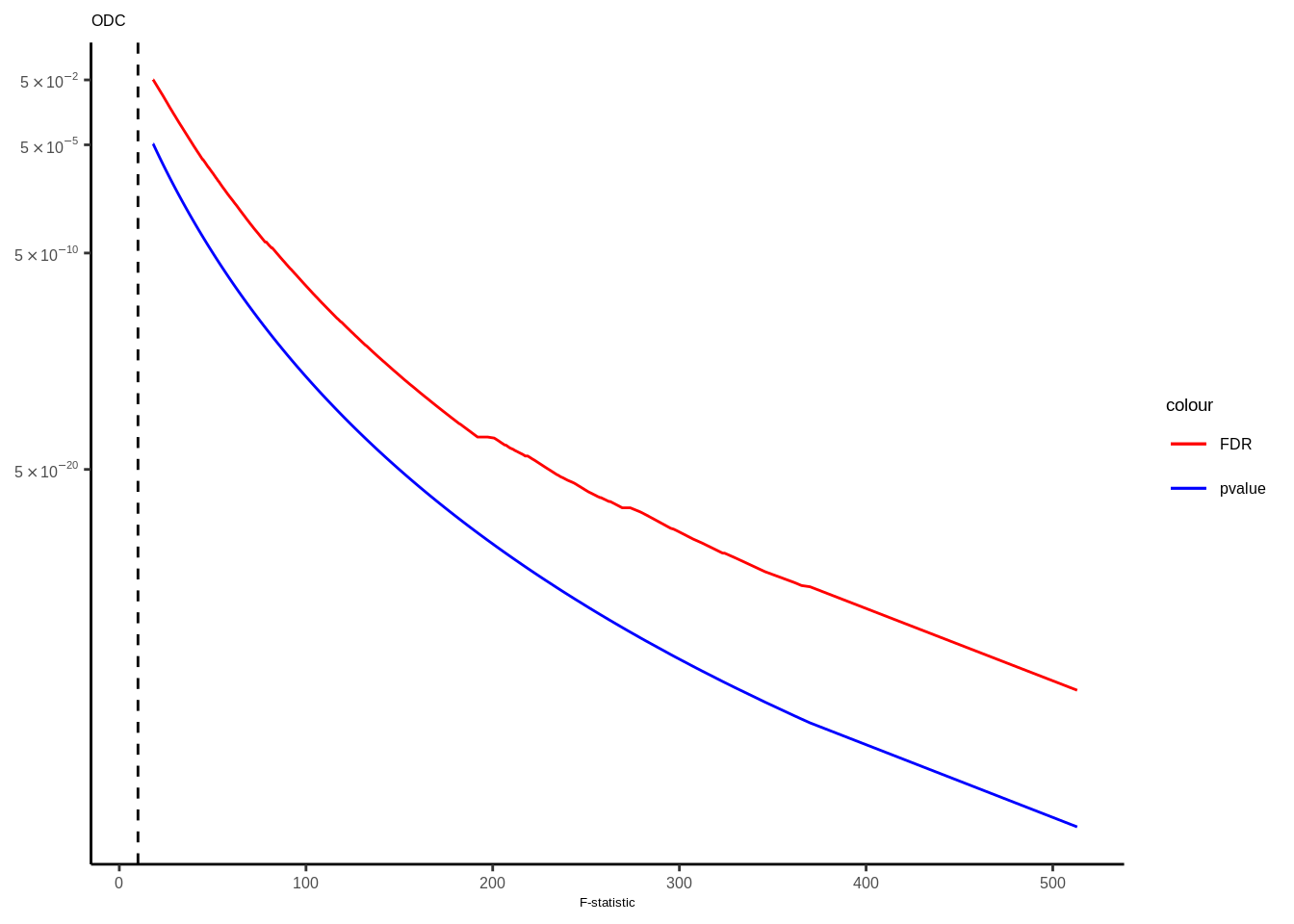
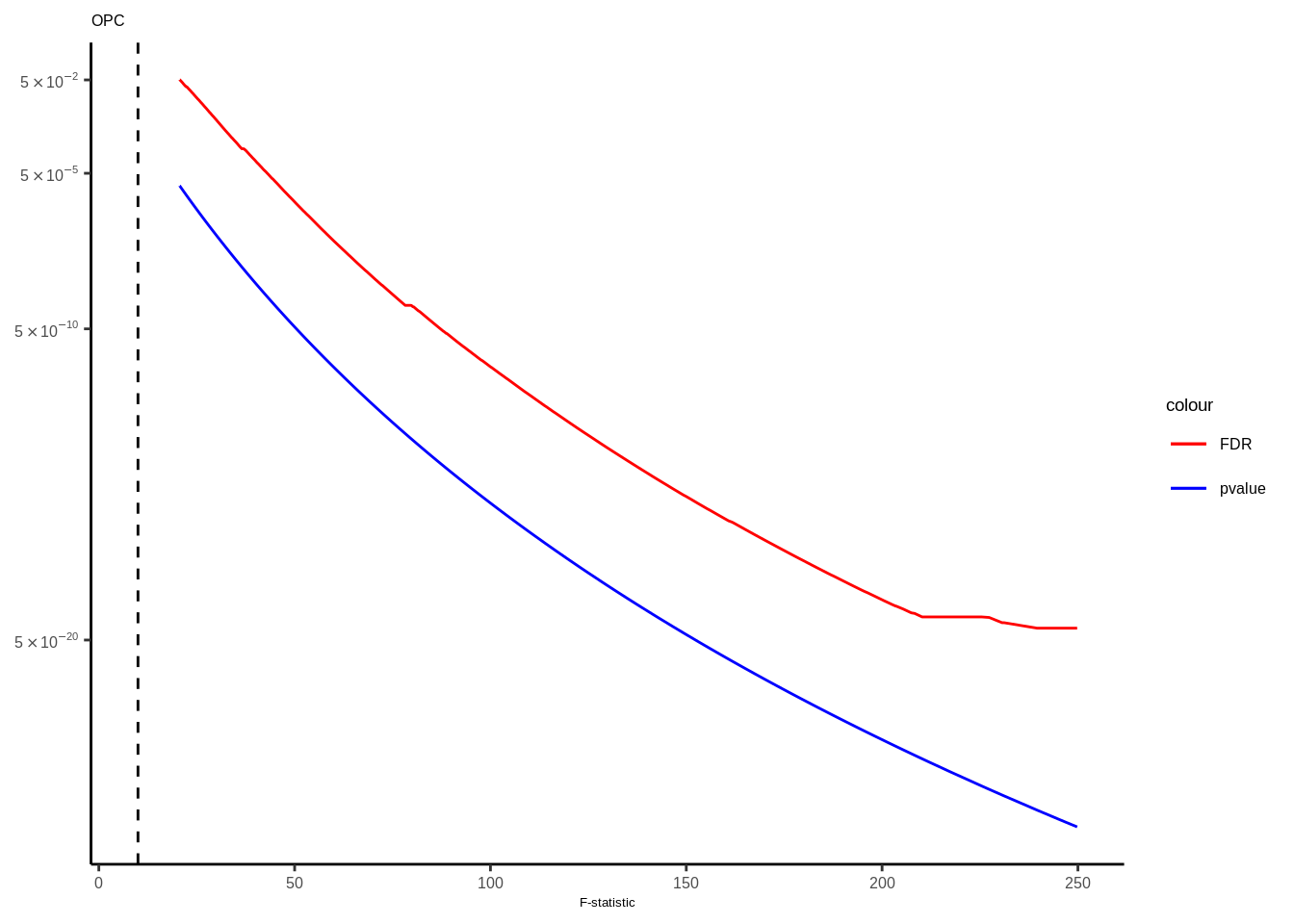
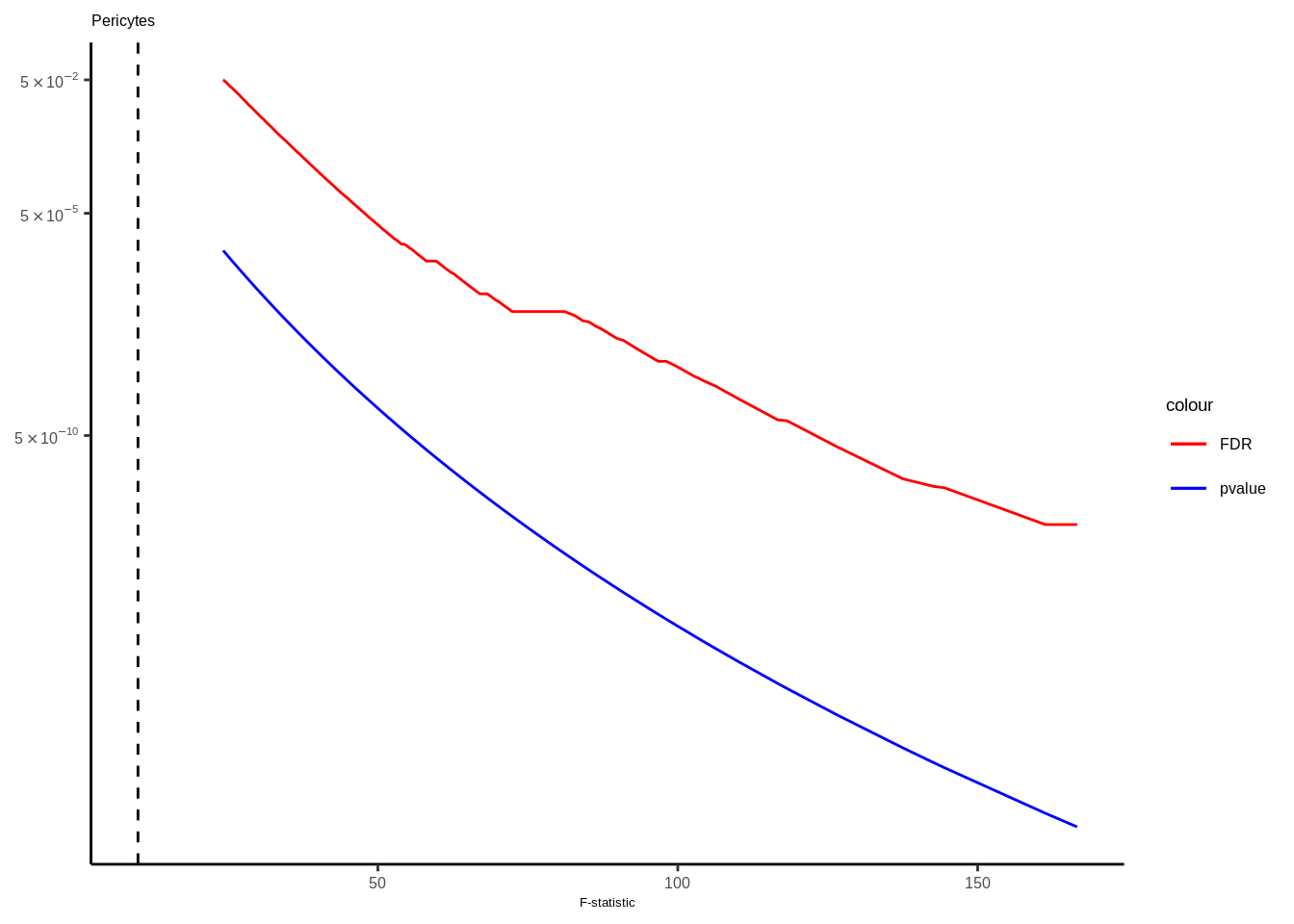
Suppl. Fig. 4
mateqtlouts<-readRDS("data/eQTL_RESULTS//mateqtlouts_0.2FDR.rds")
mateqtlouts<-lapply(mateqtlouts,function(x){
x$fstat=x$t.stat^2
return(x)})
scientific_10 <- function(x) {
parse(text=gsub("e", ".00 %*% 10^", scales::scientific_format(digits = 3)(x)))
}
library(ggplot2)
for(i in 1:length(mateqtlouts)){
tmp<-mateqtlouts[[i]]
title<-names(mateqtlouts[i])
title<-gsub("_agg_cpm","",title)
if(title=="Oligo"){
title<-"ODC"
}else if(title=="Endo"){
title<-"Endothelial"
}else if(title=="Per"){
title<-"Pericytes"
}else if(title=="Astro"){
title<-"Astrocytes"
}
tmp<-tmp[tmp$FDR<0.05,]
g<-ggplot(data=tmp,aes(x=fstat))+
geom_line(aes(y=FDR,color="FDR"),size=0.5)+
geom_line(aes(y=p.value,color="pvalue"),size=0.5)+
scale_color_manual(values = c("FDR"="red", "pvalue"="blue"))+
scale_y_continuous(trans="log10",breaks=c(5e-2,5e-5,5e-10,5e-20),labels=scientific_10)+
theme_classic()+ggtitle(title)+
xlab("F-statistic")+
geom_vline(xintercept = 10,linetype="dashed",color="black")+theme(text = element_text(family="Helvetica",size=5),axis.title.y=element_blank(),
axis.text.y=element_text(family="Helvetica",size=6),
axis.text.x=element_text(family="Helvetica",size=6),
legend.title=element_text(family="Helvetica",size=7),
legend.text=element_text(family="Helvetica",size=6))
print(g)
}
| Version | Author | Date |
|---|---|---|
| 10dd725 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| 10dd725 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| 10dd725 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| 10dd725 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| 10dd725 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| 10dd725 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| 10dd725 | Alexander Haglund | 2022-12-01 |
Suppl. Fig. 5
Suppl. Fig. 5a
full<-read.table("data/COLOC_MR_RESULTS//2022-10-25_FULL_MR_RES.txt")
full$celltype_gene<-paste0(full$celltype,".",full$gene)
scz<-full[full$GWAS=="SCZ",]
iq<-full[full$GWAS=="IQ",]
common<-intersect(iq$celltype_gene,scz$celltype_gene)
#remove ct/gene combinations in other traits - we are only interested in the SCZ/IQ overlap
filtered<-full[full$celltype_gene %in% common,]
filtered<-filtered[!filtered$GWAS %in% c("SCZ","IQ"),]
to_exclude<-unique(filtered$celltype_gene)
common<-common[!common %in% to_exclude]
#filter both
scz<-scz[match(common,scz$celltype_gene),]
iq<-iq[match(common,iq$celltype_gene),]
df<-data.frame(celltype_gene=common,IQ=iq$IVW_beta,SCZ=scz$IVW_beta)
##now plot
ylims=c(-0.2,0.2)
xlims=c(-0.7,0.7)
g<-ggplot(df,aes(x=SCZ,y=IQ,fill=celltype_gene))+
geom_point(shape=21,size=2)+
scale_y_continuous(limits=ylims)+
scale_x_continuous(limits=xlims)+
geom_vline(xintercept =0,linetype="dashed",size=0.3)+
geom_hline(yintercept =0,linetype="dashed",size=0.3)+theme_classic()+
theme(text=element_text(size=5,family="Helvetica"),legend.title=element_blank(),
axis.text.y=element_text(size=5,family="Helvetica"),axis.text.x=element_text(size=5,family="Helvetica"),
legend.text = element_text(family="Helvetica",size=5,face="bold"),legend.spacing.y = unit(-1, 'cm'))+
xlab("SCZ - MR beta")+ylab("IQ - MR beta")+
guides(fill = guide_legend(byrow = TRUE))
g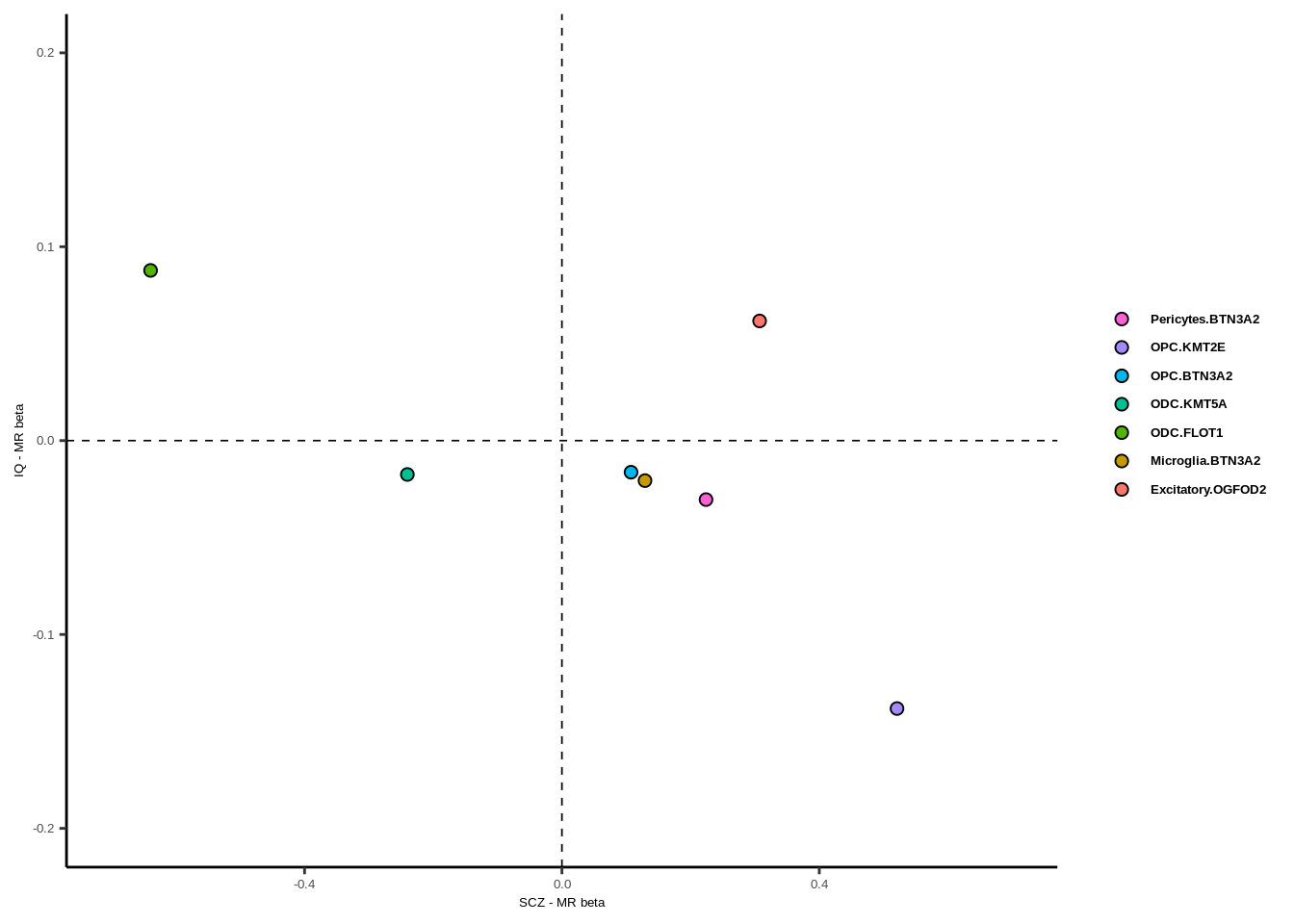
| Version | Author | Date |
|---|---|---|
| 0f7883c | Alexander Haglund | 2022-12-01 |
Suppl. Fig. 5b
full<-read.table("data/COLOC_MR_RESULTS//2022-10-25_FULL_MR_RES.txt")
full$celltype_gene<-paste0(full$celltype,".",full$gene)
scz<-full[full$GWAS=="SCZ",]
neur<-full[full$GWAS=="NEUR",]
common<-intersect(neur$celltype_gene,scz$celltype_gene)
filtered<-full[full$celltype_gene %in% common,]
filtered<-filtered[!filtered$GWAS %in% c("SCZ","NEUR"),]
to_exclude<-unique(filtered$celltype_gene)
common<-common[!common %in% to_exclude]
#filter both
scz<-scz[match(common,scz$celltype_gene),]
neur<-neur[match(common,neur$celltype_gene),]
df<-data.frame(celltype_gene=common,NEUR=neur$IVW_beta,SCZ=scz$IVW_beta)
##now plot
ylims=c(-0.2,0.2)
xlims=c(-0.8,0.8)
g<-ggplot(df,aes(x=SCZ,y=NEUR,fill=celltype_gene))+
geom_point(shape=21,size=2)+
scale_y_continuous(limits=ylims)+
scale_x_continuous(limits=xlims)+
geom_vline(xintercept =0,linetype="dashed",size=0.3)+
geom_hline(yintercept =0,linetype="dashed",size=0.3)+theme_classic()+
theme(text=element_text(size=5,family="Helvetica"),legend.title=element_blank(),
axis.text.y=element_text(size=5,family="Helvetica"),axis.text.x=element_text(size=5,family="Helvetica"),
legend.text = element_text(family="Helvetica",size=5,face="bold"),legend.spacing.y = unit(-1, 'cm'))+
xlab("SCZ - MR beta")+ylab("NEUR - MR beta")+guides(fill = guide_legend(byrow = TRUE))
g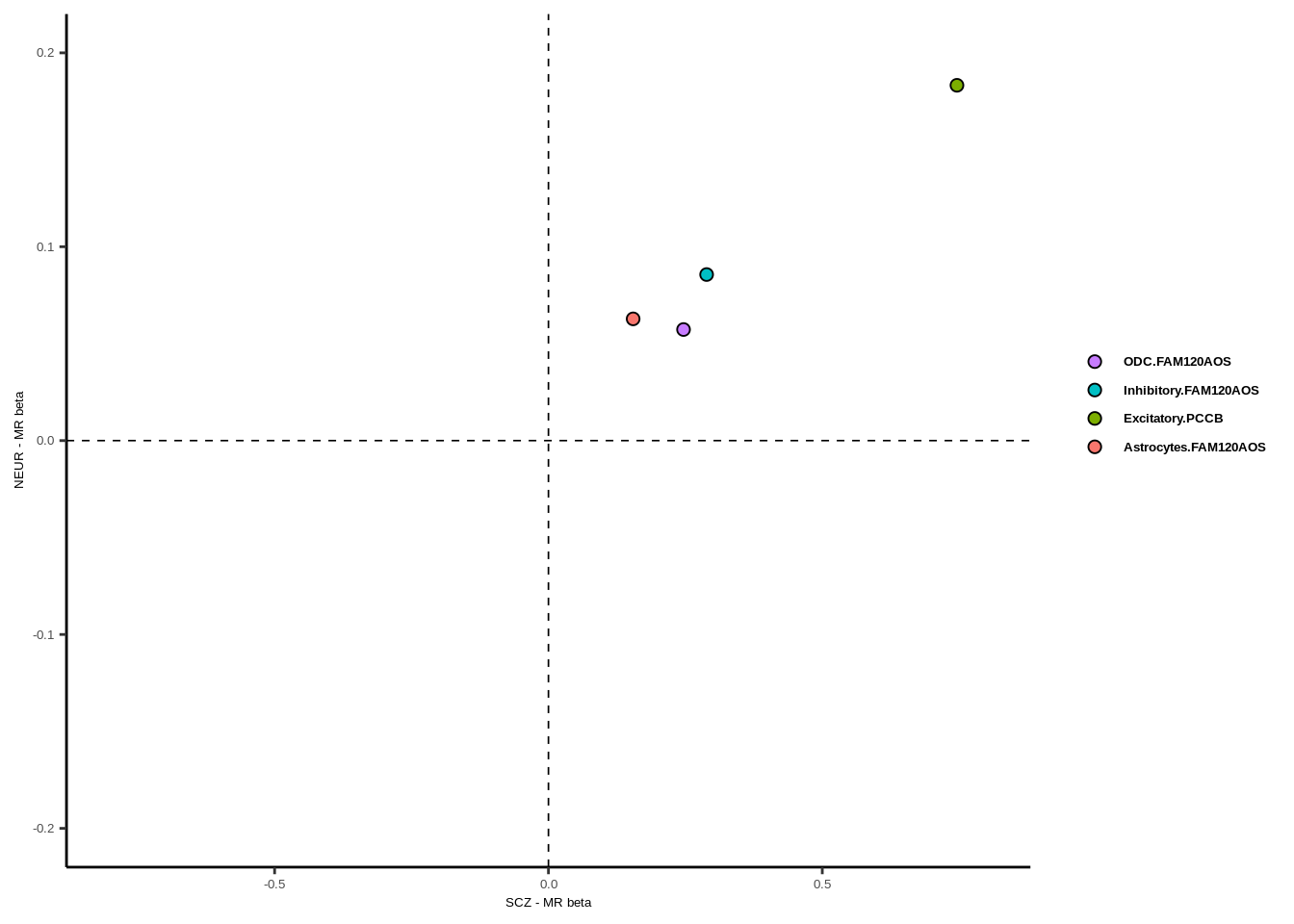
| Version | Author | Date |
|---|---|---|
| 0f7883c | Alexander Haglund | 2022-12-01 |
Suppl. Fig. 5c
full<-read.table("data/COLOC_MR_RESULTS//2022-10-25_FULL_MR_RES.txt")
full<-full[full$IVW<0.05,]
tmp<-full
df<-data.frame()
for(i in 1:length(unique(tmp$GWAS))){
gwas<-unique(tmp$GWAS)[i]
tmp_df<-tmp[grep(gwas,tmp$GWAS),]
cellvec<-as.vector(table(tmp_df$celltype))
df<-rbind(df,data.frame(gwas=gwas,celltype=names(table(tmp_df$celltype)),occurrence=cellvec))
}
g<-ggplot(data=df,aes(y=occurrence,x=gwas,fill=celltype))+geom_bar(stat="identity",colour="black")
g<-g+geom_text(aes(label=occurrence),family="Helvetica",size=5*0.36, position = position_stack(vjust = 0.5))+
scale_fill_manual(values=color_pal)+theme_classic()+
scale_y_continuous(expand = c(0, 0))+
theme(axis.text.x=element_text(size=5,face="bold",angle=45,vjust=0.01),
axis.text.y=element_text(size=5,face="bold"),
axis.title.y=element_text(size=7,face="bold",margin=margin(r=20)),
axis.title.x=element_text(size=7,face="bold",margin=margin(t=20)),
legend.key.size=unit(0.7,"cm"),
legend.text=element_text(size=5),
title=element_text(size=7,face="bold"),
panel.background = element_rect(fill='transparent'), #transparent panel bg
plot.background = element_rect(fill='transparent', color=NA), #transparent plot bg
panel.grid.major = element_blank(), #remove major gridlines
panel.grid.minor = element_blank(), #remove minor gridlines
legend.background = element_rect(fill='transparent'), #transparent legend bg
legend.box.background = element_rect(fill='transparent'))#transparent legend panel
g<-g+theme(text=element_text(family="Helvetica"),
axis.title.x=element_blank(),
axis.title.y=element_blank(), axis.text.x=element_text(size=5,angle=45),
legend.position = "none")+scale_y_continuous(expand = c(0, 0))Scale for 'y' is already present. Adding another scale for 'y', which will
replace the existing scale.g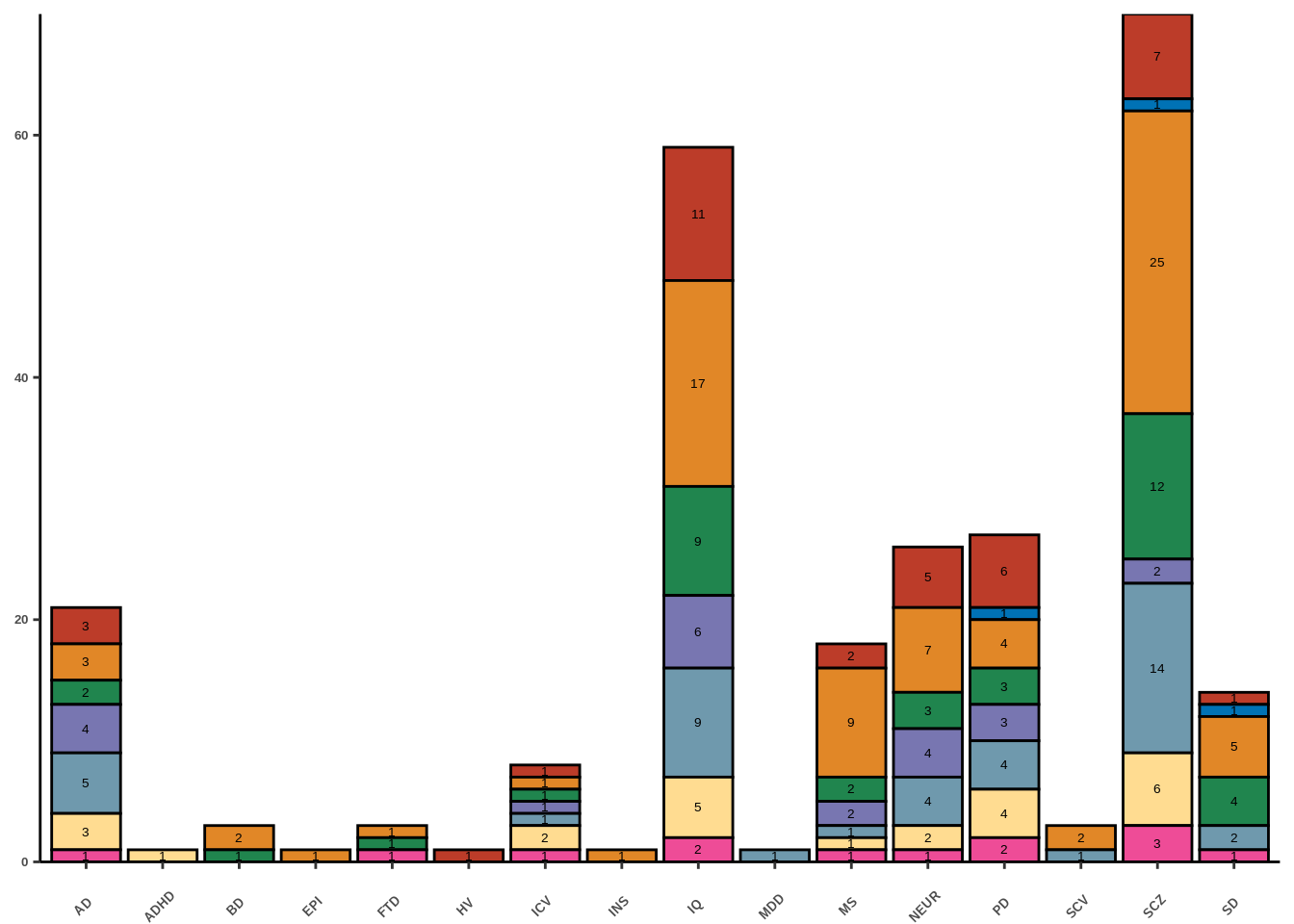
| Version | Author | Date |
|---|---|---|
| 0f7883c | Alexander Haglund | 2022-12-01 |
Suppl. Fig. 5d
full<-read.table("data/COLOC_MR_RESULTS//2022-10-25_FULL_MR_RES.txt")
full<-full[full$IVW<0.05,]
full$trait_gene<-paste0(full$GWAS,"_",full$gene)
tmp<-full[full$GWAS=="AD",]
tmp<-tmp[order(tmp$celltype),]
tmp$gene<-factor(tmp$gene,levels=unique(tmp$gene))
color_pal=ggsci::pal_nejm("default")(8)
colorvec<-c(Astrocytes=color_pal[1],
Endothelial=color_pal[2],
Excitatory=color_pal[3],
Inhibitory=color_pal[4],Microglia=color_pal[5],
ODC=color_pal[6],OPC=color_pal[7],Pericytes=color_pal[8])
g<-ggplot(tmp,aes(y=gene,x=IVW_beta,fill=celltype))+
geom_point(colour="black",shape=21,stroke=0.3,size=2)+
scale_fill_manual(values = colorvec)+
theme_classic()+
geom_vline(xintercept=0,linetype="dashed",size=0.3)+scale_x_continuous(limits=c(-0.6,0.6))
g2<-g+
theme(text=element_text(family="Helvetica",size=5),legend.text = element_text(family="Helvetica",size=5,face="bold"),
legend.title=element_blank(),
legend.spacing.y = unit(-1, 'cm'),axis.text.y=element_text(family="Helvetica",size=5,face="bold"))+guides(fill = guide_legend(byrow = TRUE))
g2
| Version | Author | Date |
|---|---|---|
| 0f7883c | Alexander Haglund | 2022-12-01 |
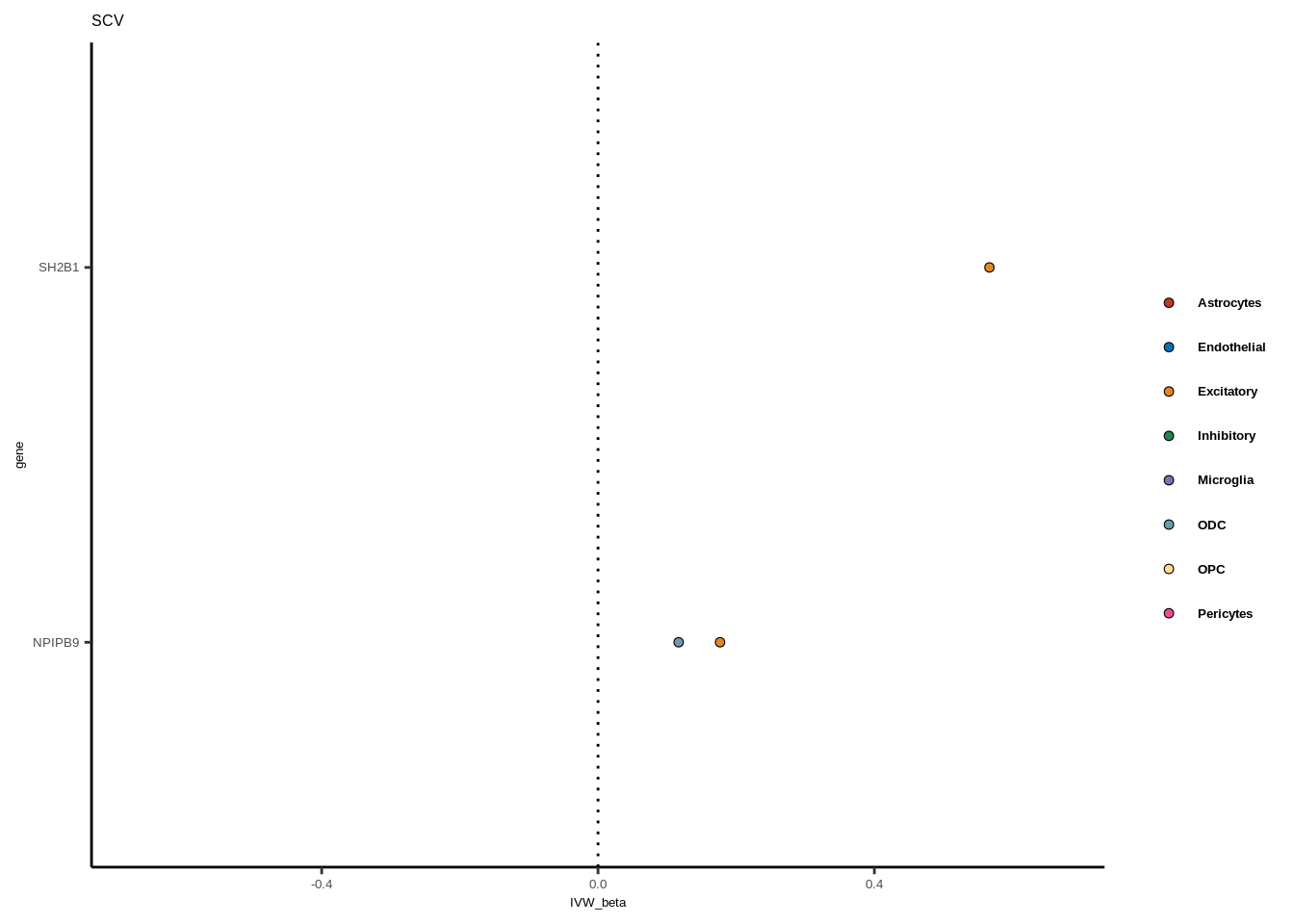
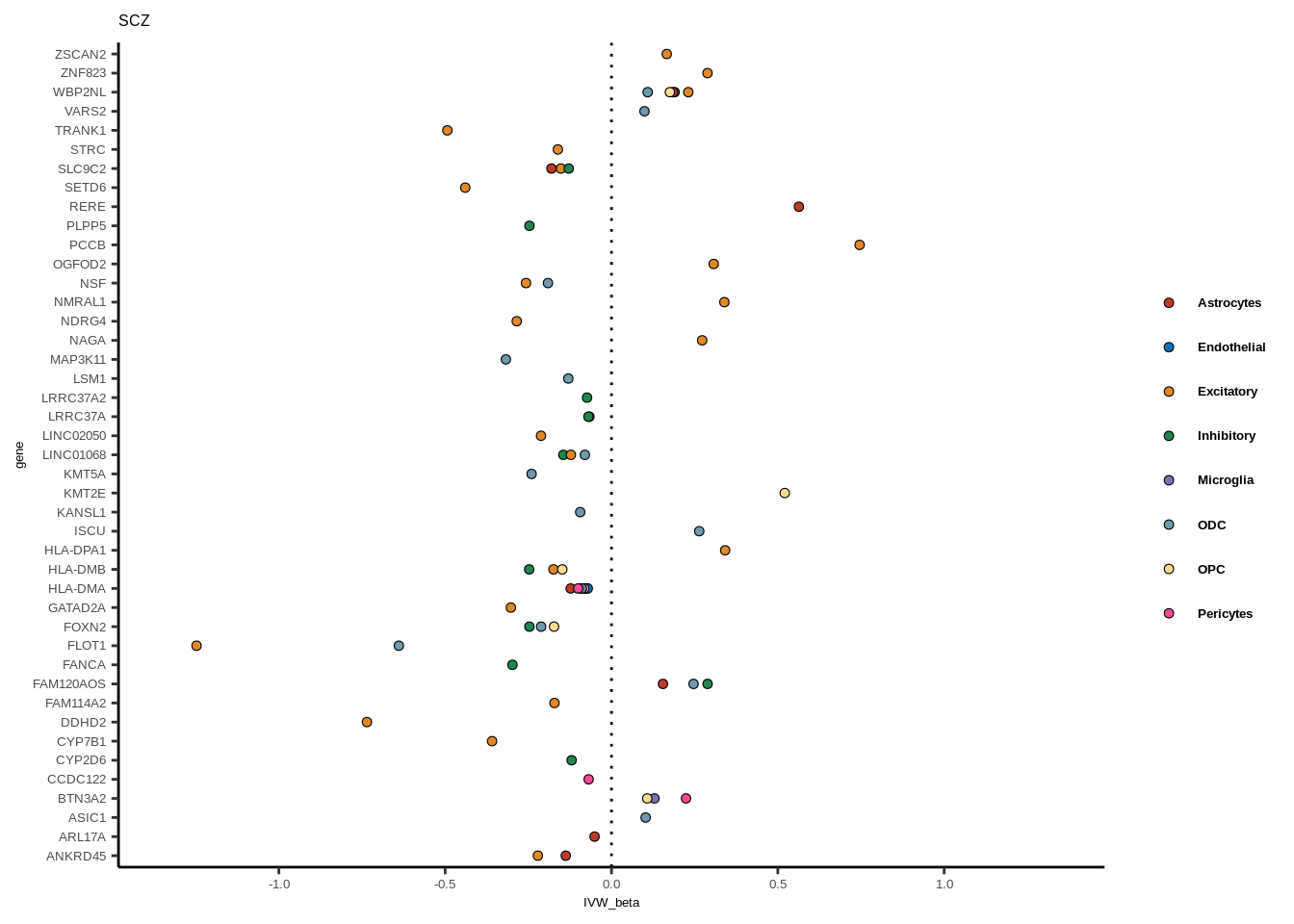
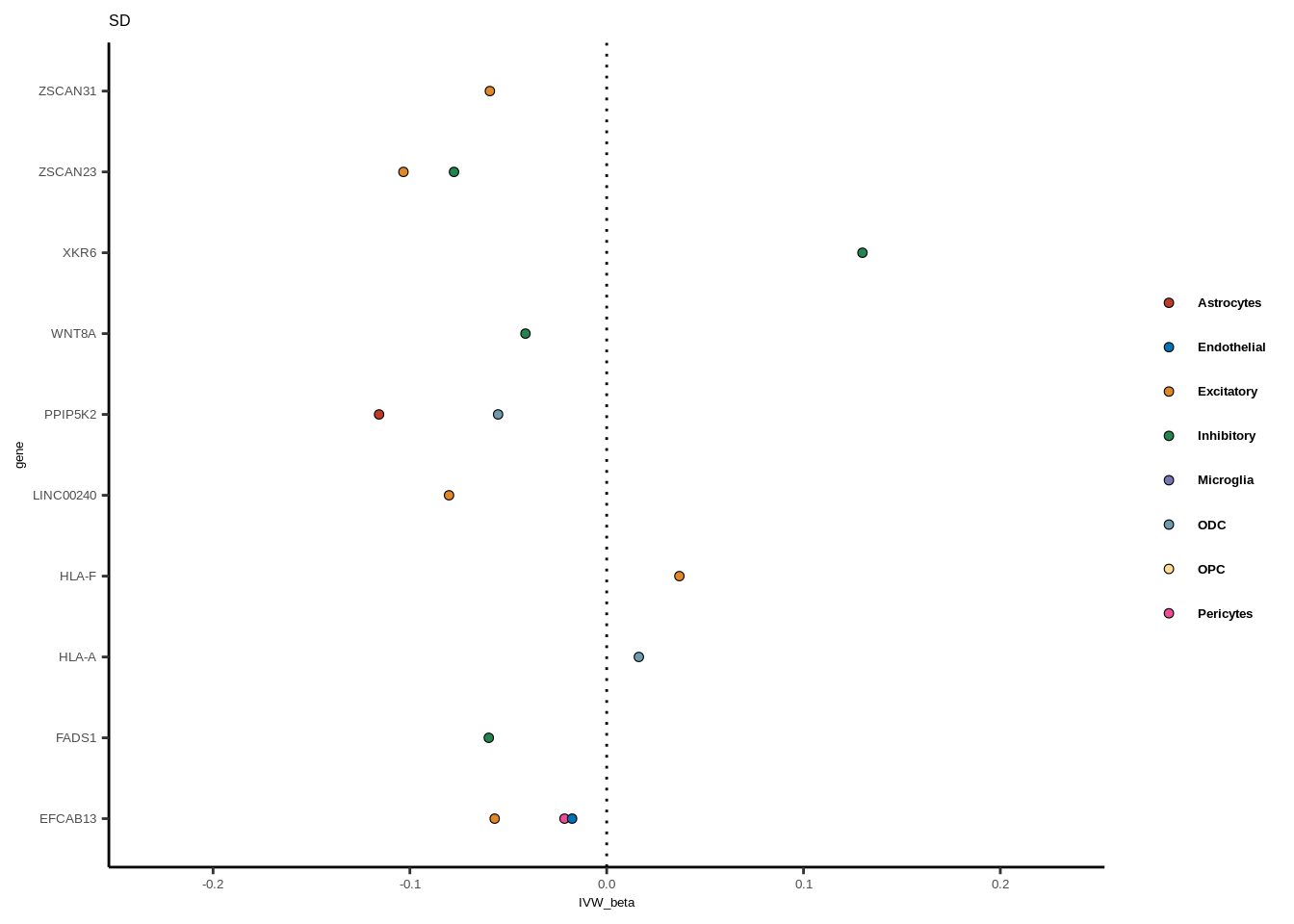
Suppl. Fig. 6
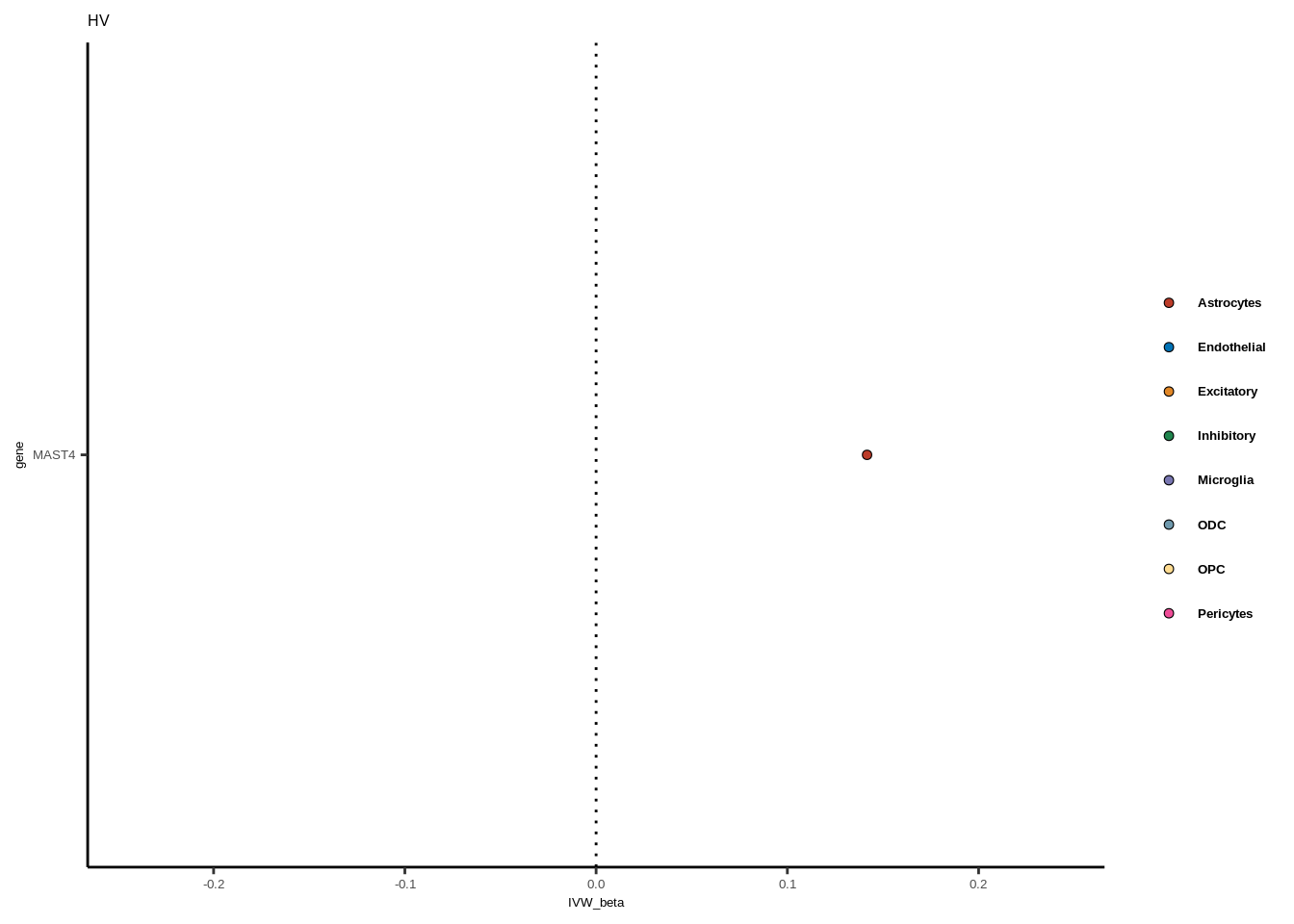
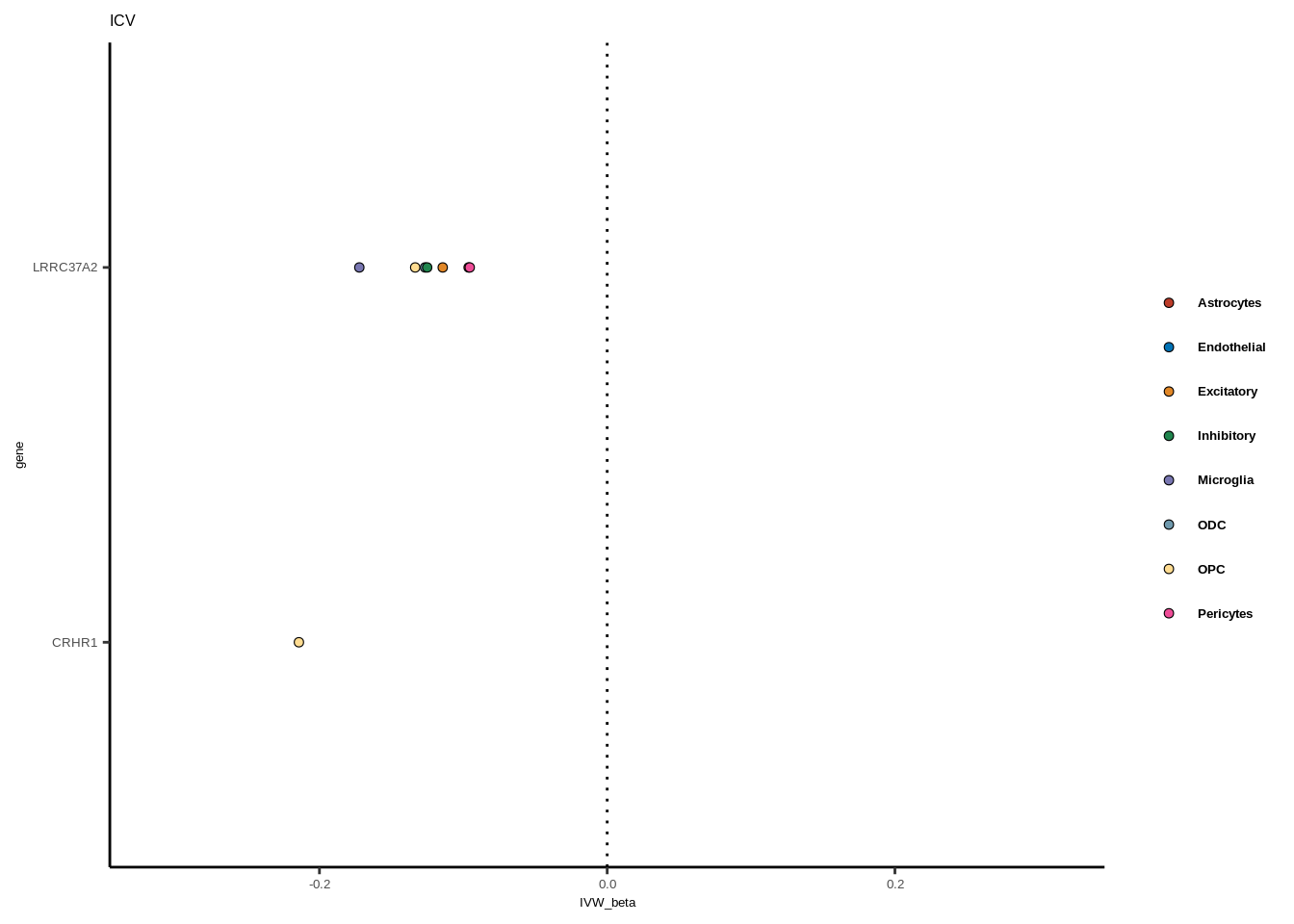
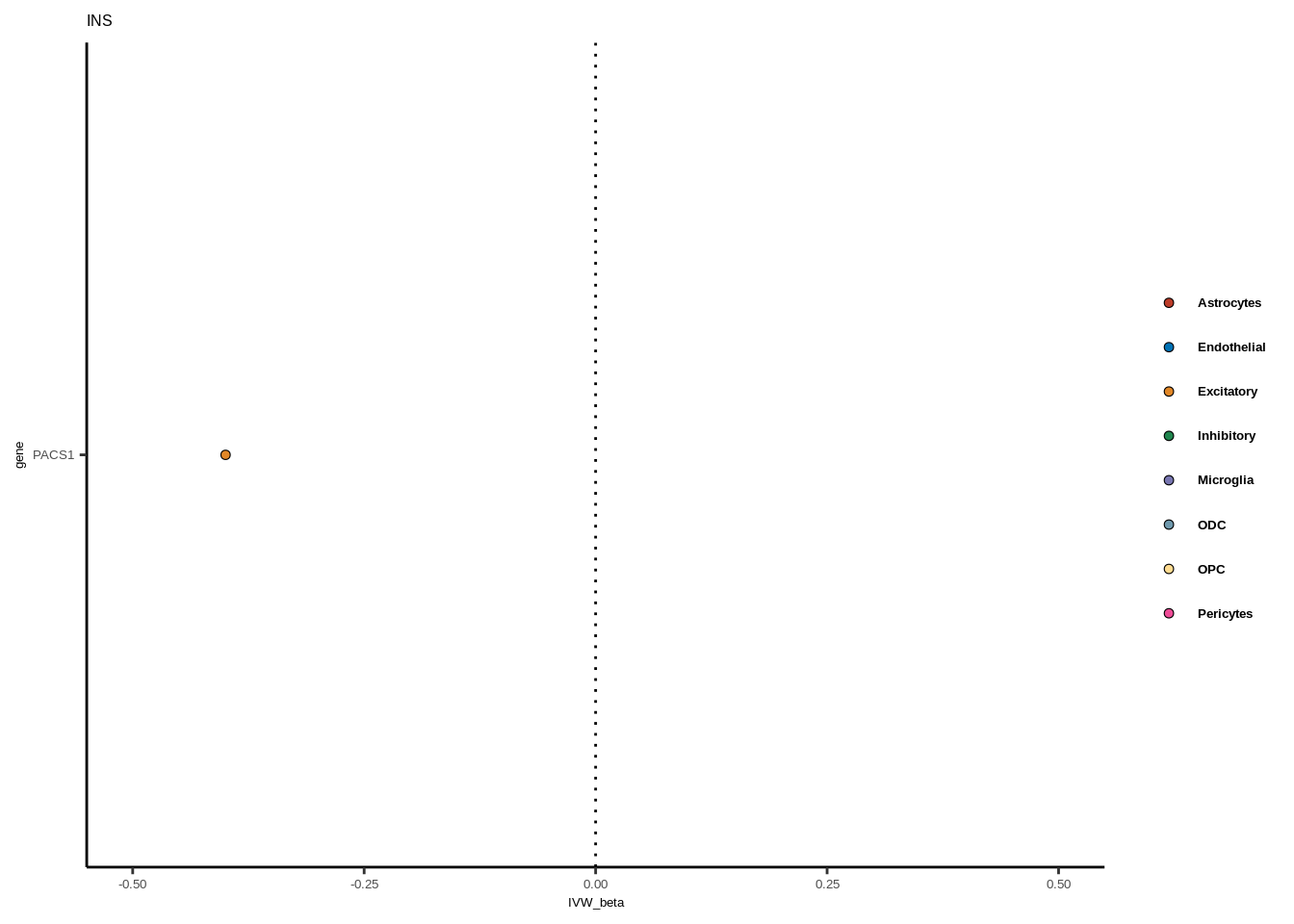
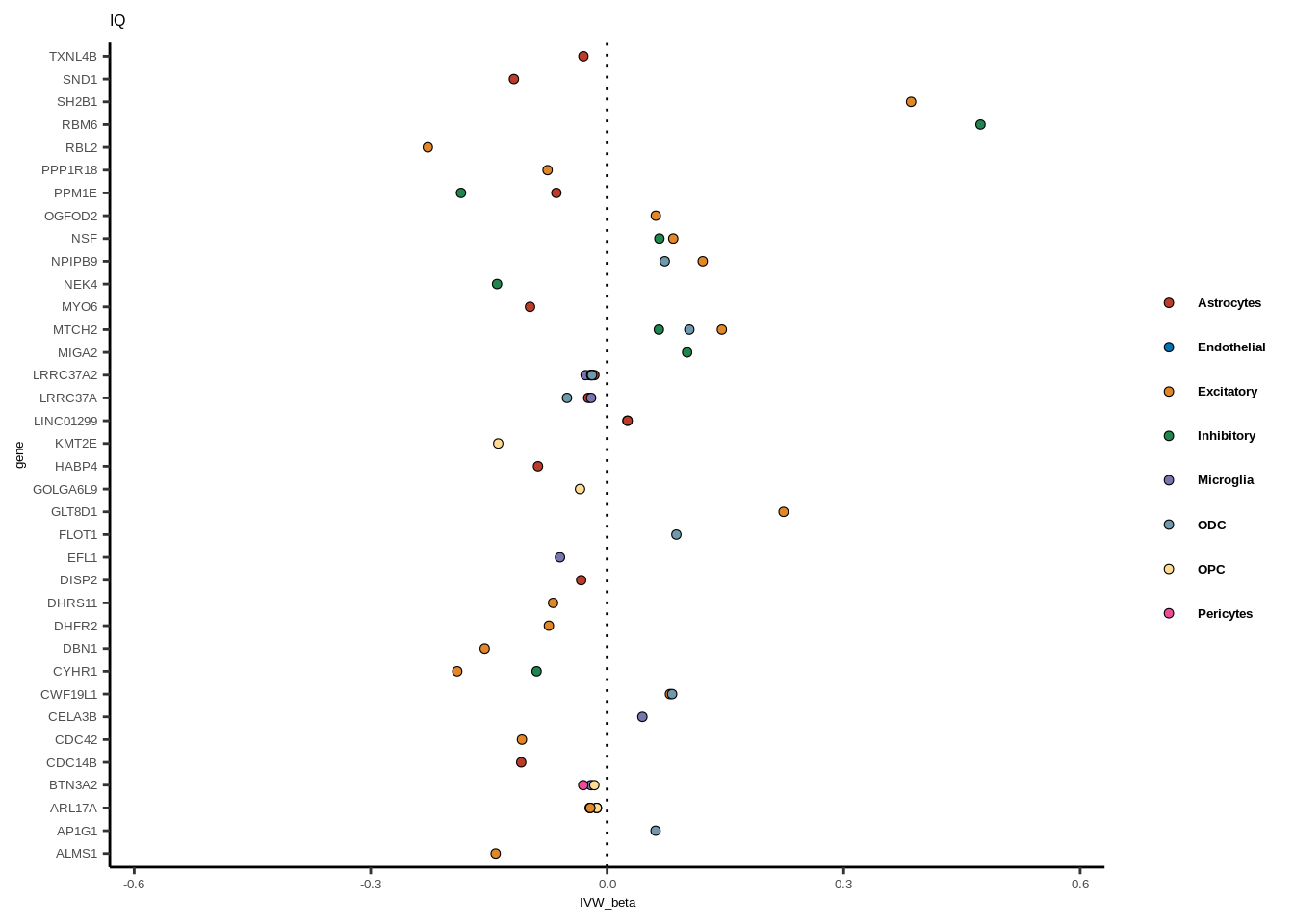
full<-read.table("data/COLOC_MR_RESULTS//2022-10-25_FULL_MR_RES.txt")
full<-full[full$IVW<0.05,]
full$trait_gene<-paste0(full$GWAS,"_",full$gene)
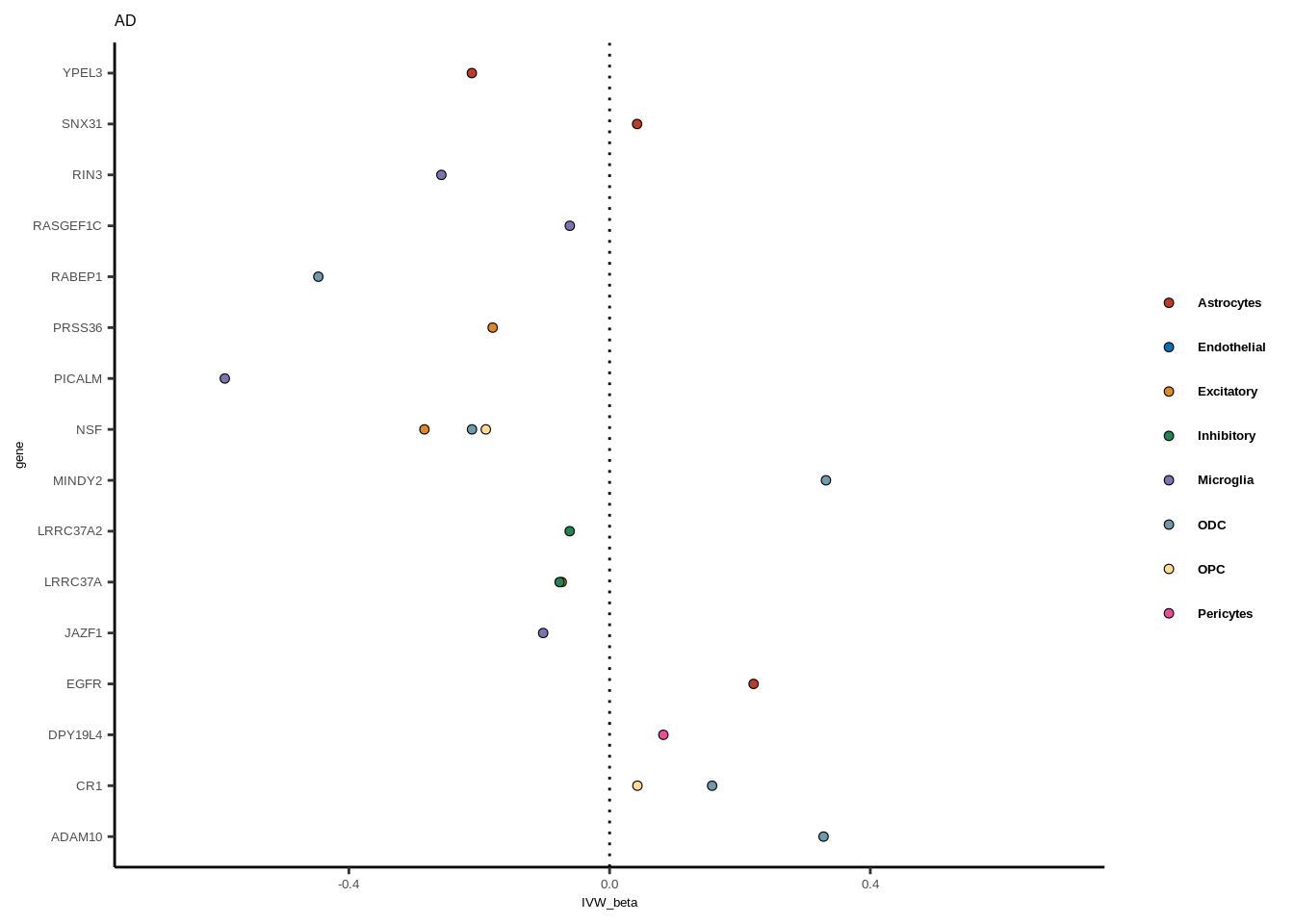
tmp<-full[full$GWAS=="AD",]
tmp<-tmp[order(tmp$celltype),]
tmp$gene<-factor(tmp$gene,levels=unique(tmp$gene))
color_pal=ggsci::pal_nejm("default")(8)
colorvec<-c(Astrocytes=color_pal[1],
Endothelial=color_pal[2],
Excitatory=color_pal[3],
Inhibitory=color_pal[4],Microglia=color_pal[5],
ODC=color_pal[6],OPC=color_pal[7],Pericytes=color_pal[8])
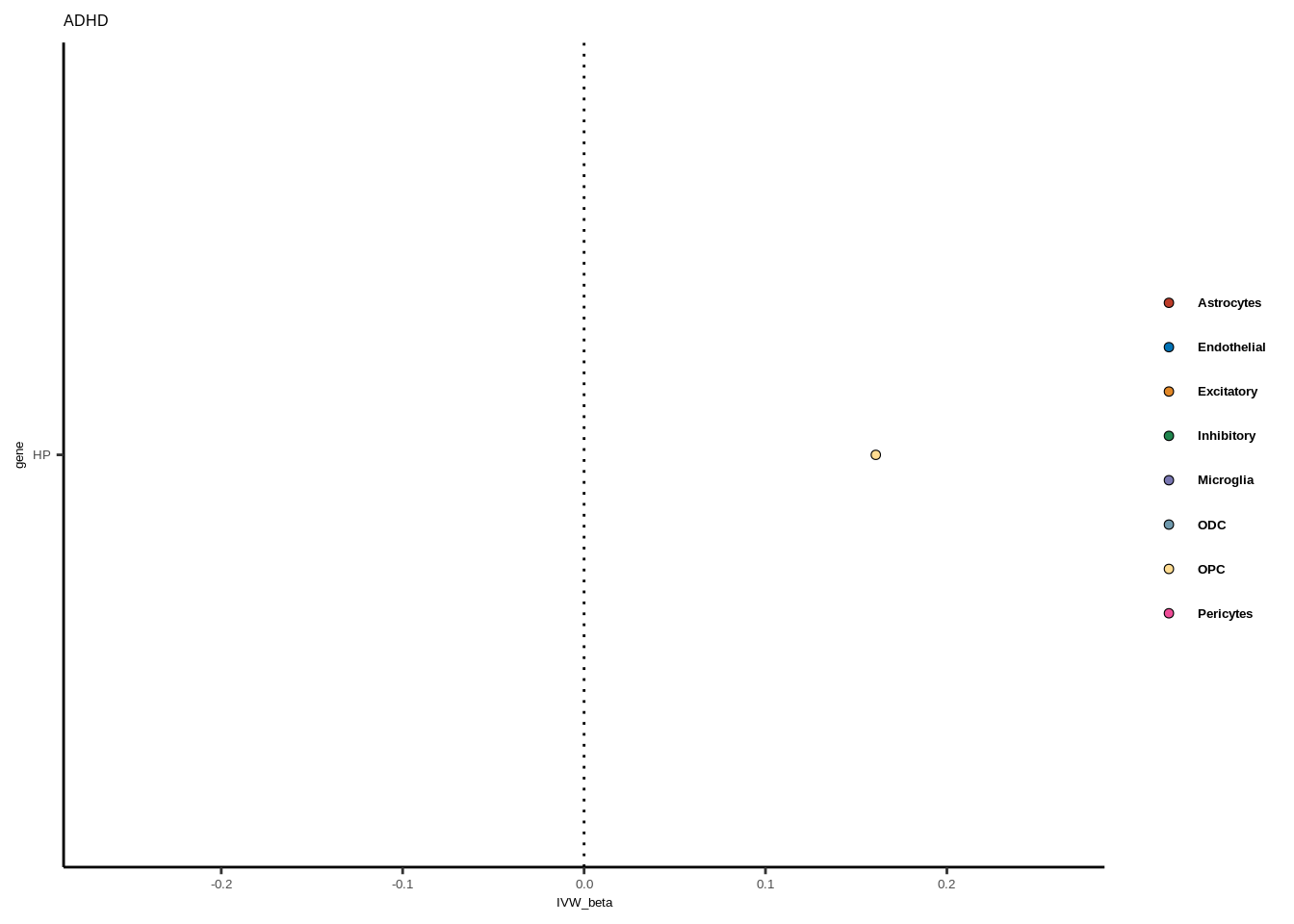
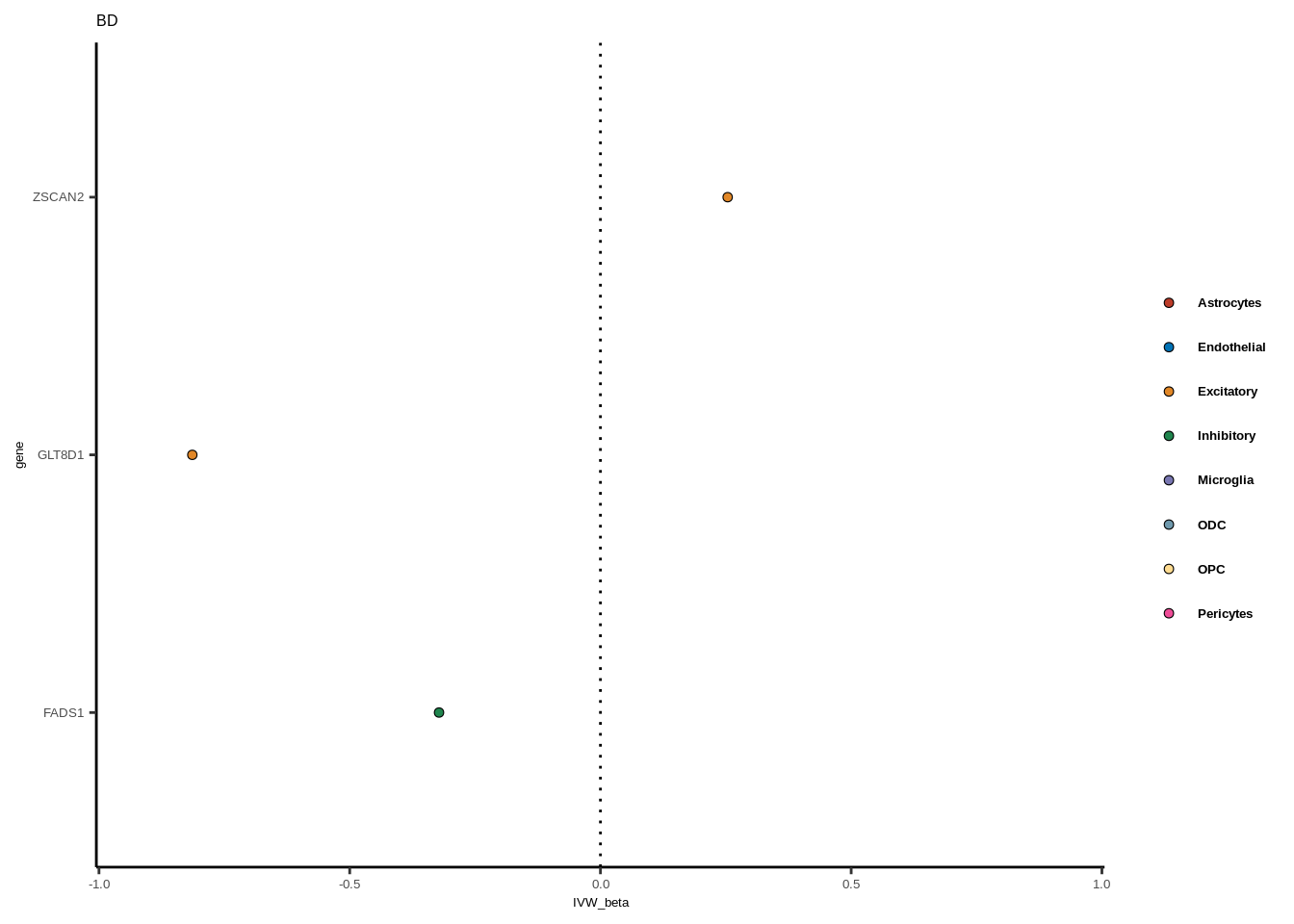
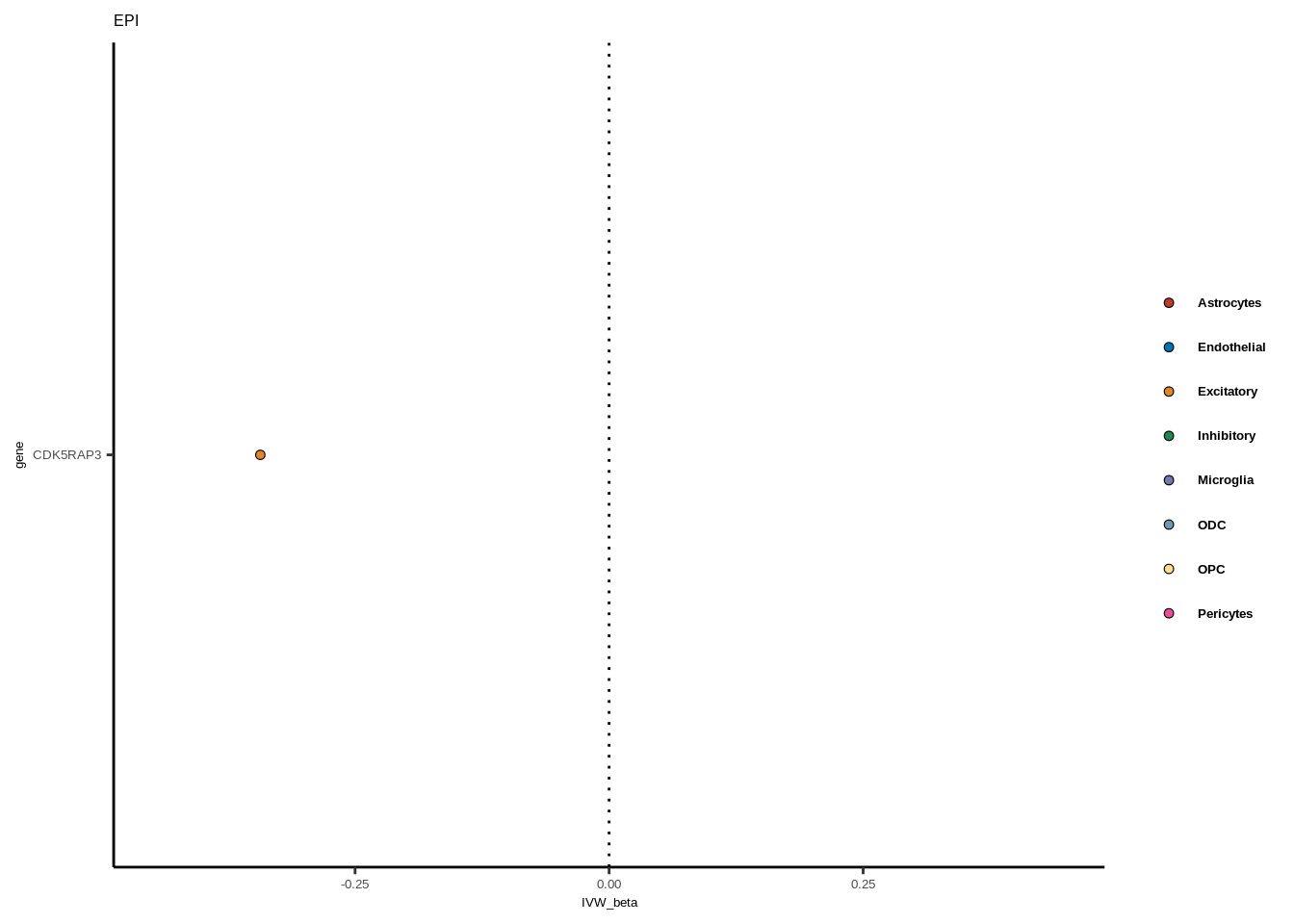
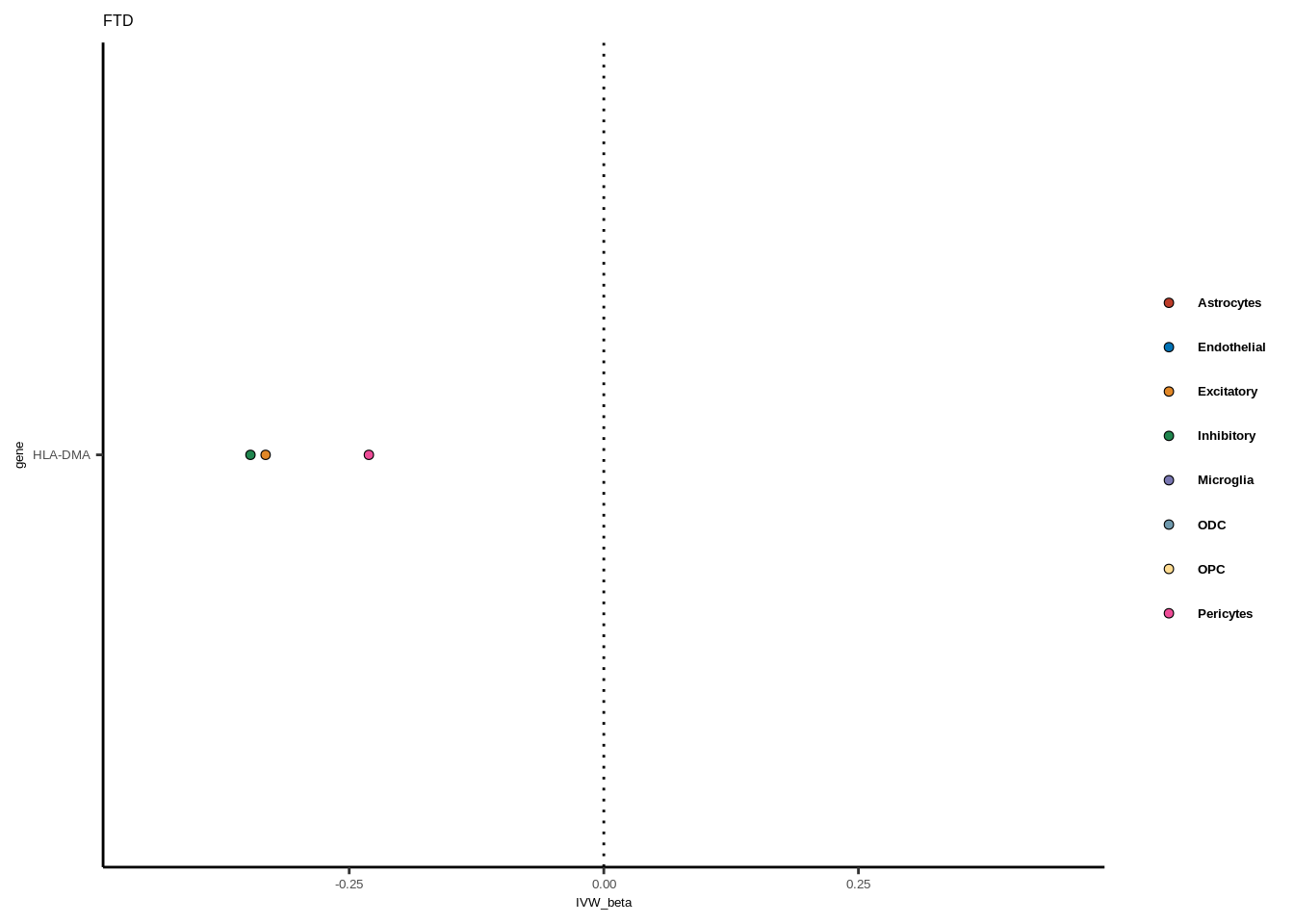
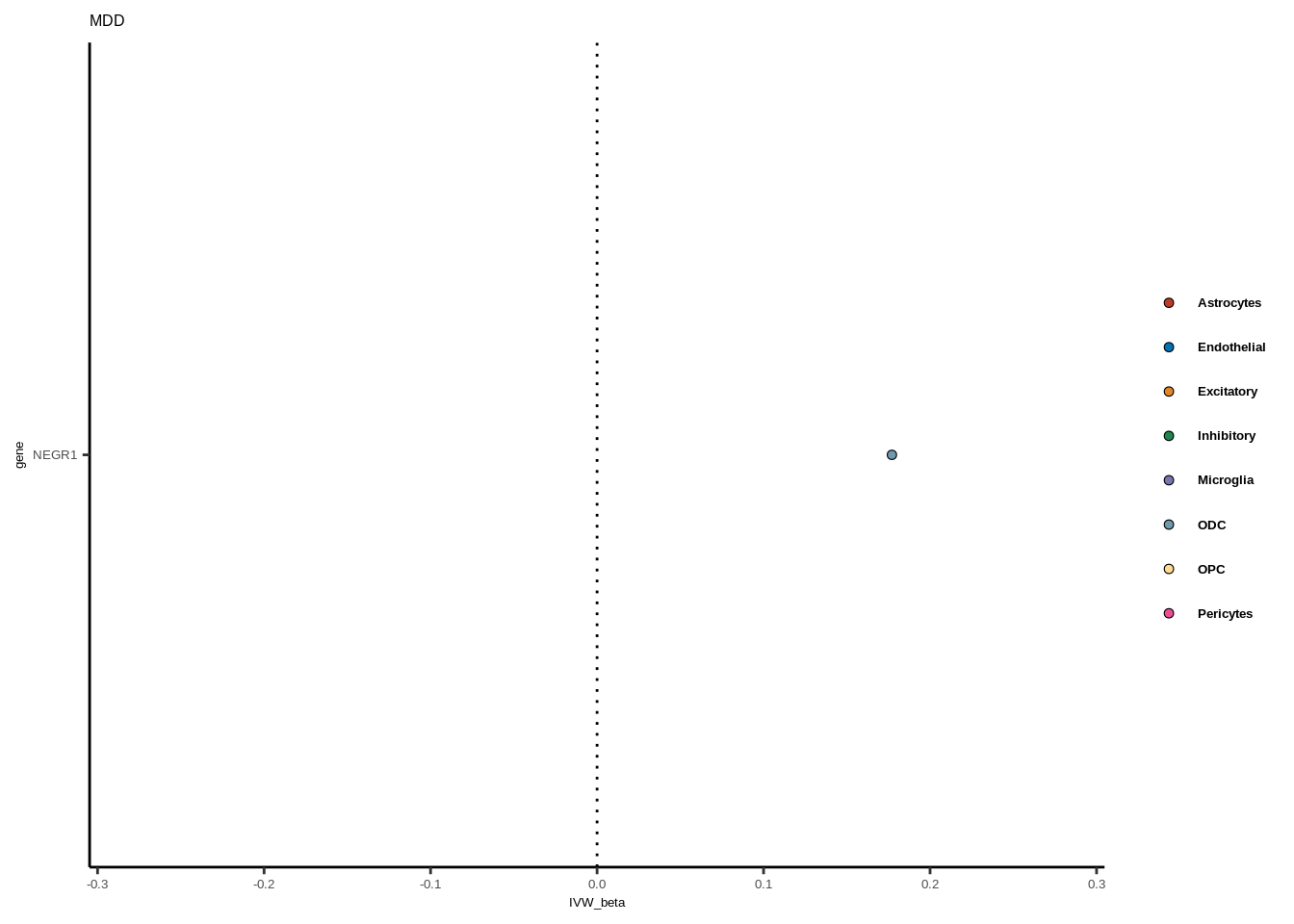
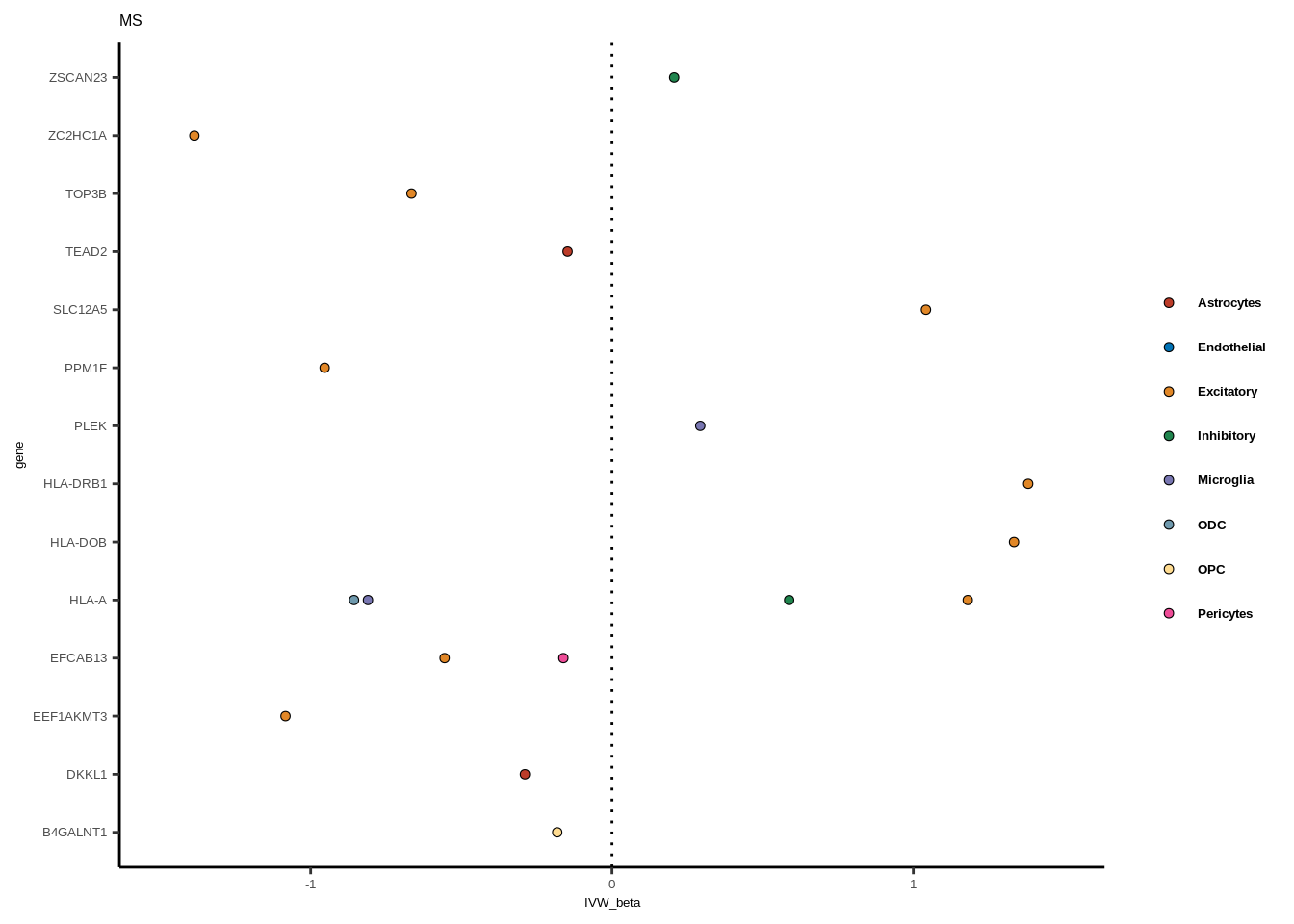
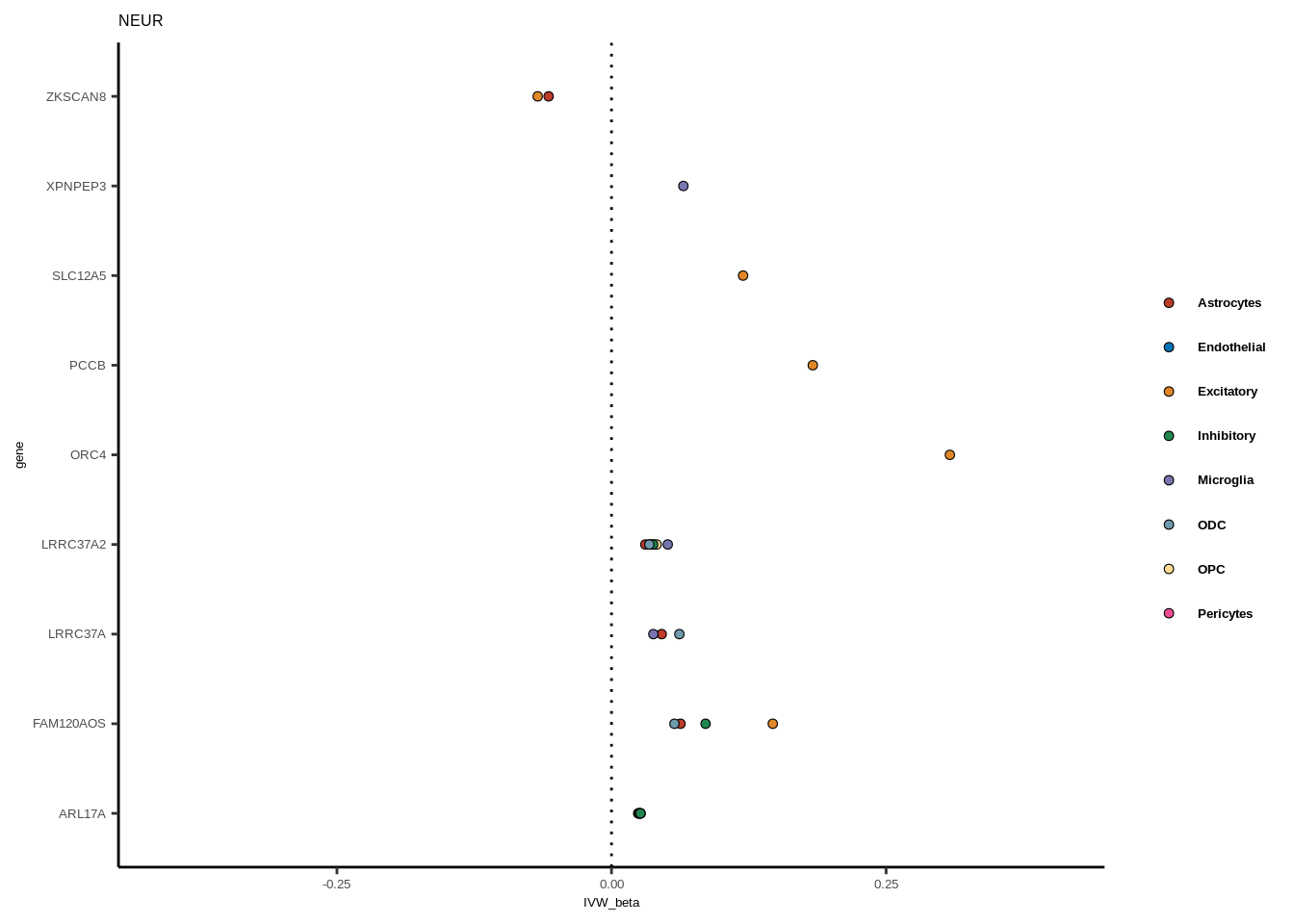
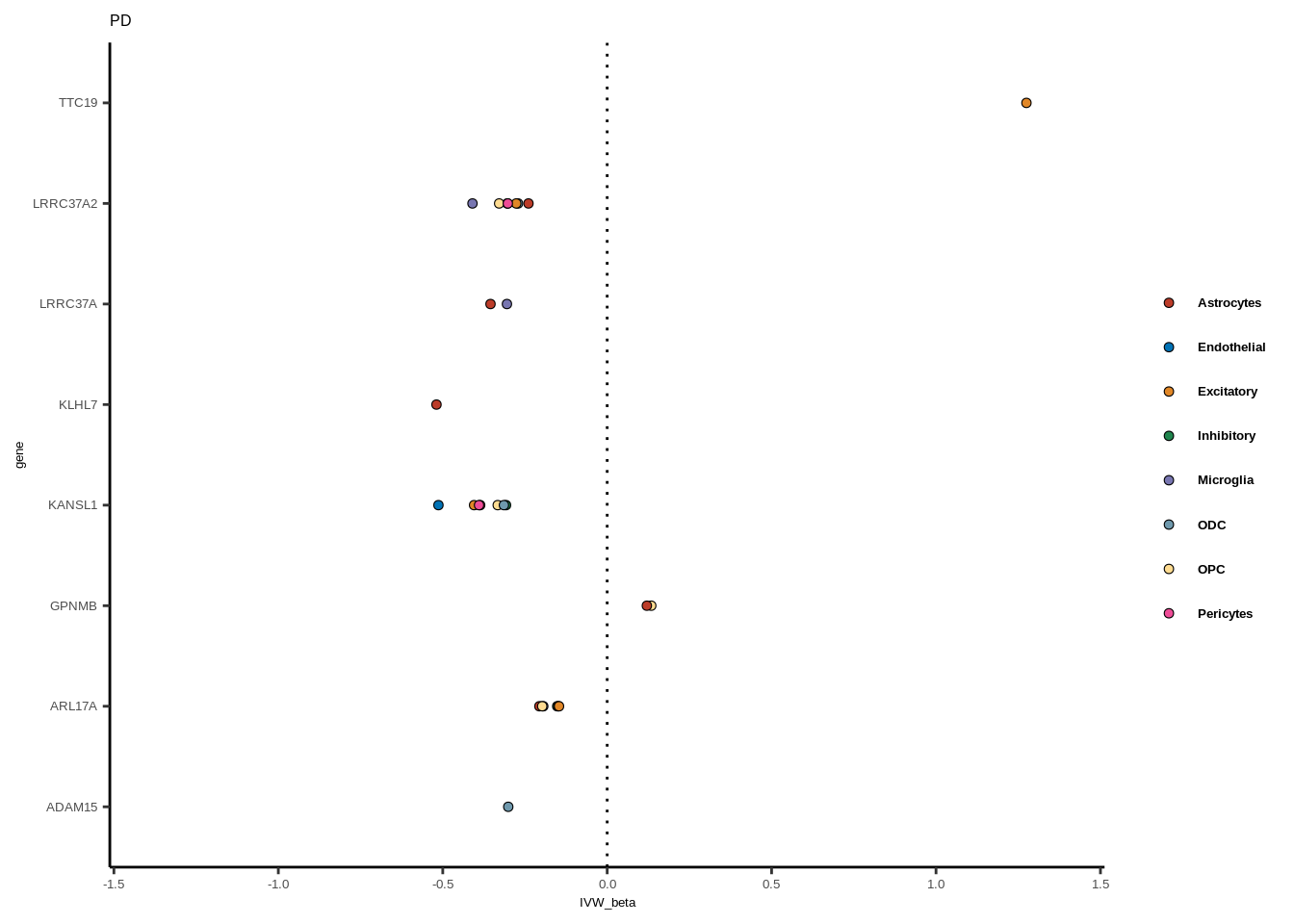
for(i in 1:length(unique(full$GWAS))){
gwas<-unique(full$GWAS)[i]
tmp<-full[full$GWAS %in% gwas,]
len<-nrow(tmp)
fig_height=3/20*len
lims=max(abs(tmp$IVW_beta))+0.1
g<-ggplot(tmp,aes(y=gene,x=IVW_beta,fill=celltype))+
geom_point(colour="black",shape=21,stroke=0.3)+
scale_fill_manual(values = colorvec)+
theme_classic()+
geom_vline(xintercept=0,linetype="dotted")+scale_x_continuous(limits=c(lims*-1,lims))
g<-g+
theme(text=element_text(size=5,family="Helvetica"),legend.title=element_blank(),
axis.text.y=element_text(size=5,family="Helvetica"),axis.text.x=element_text(size=5,family="Helvetica"),
legend.text = element_text(family="Helvetica",size=5,face="bold"))+ggtitle(gwas)
print(g)
}
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
| Version | Author | Date |
|---|---|---|
| dbf14b0 | Alexander Haglund | 2022-12-01 |
sessionInfo()R version 4.0.5 (2021-03-31)
Platform: x86_64-conda-linux-gnu (64-bit)
Running under: Ubuntu 18.04.6 LTS
Matrix products: default
BLAS/LAPACK: /home/ah3918/anaconda3/envs/ODIN3/lib/libopenblasp-r0.3.12.so
locale:
[1] LC_CTYPE=en_GB.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB.UTF-8 LC_COLLATE=en_GB.UTF-8
[5] LC_MONETARY=en_GB.UTF-8 LC_MESSAGES=en_GB.UTF-8
[7] LC_PAPER=en_GB.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_GB.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] ggrepel_0.9.1 ComplexUpset_1.3.3 UpSetR_1.4.0 reshape_0.8.9
[5] tidyr_1.2.1 cowplot_1.1.1 dplyr_1.0.9 ggsci_2.9
[9] viridis_0.6.2 viridisLite_0.4.1 ggplot2_3.3.6 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] tidyselect_1.1.2 xfun_0.32 bslib_0.4.0 purrr_0.3.4
[5] colorspace_2.0-3 vctrs_0.5.0 generics_0.1.3 htmltools_0.5.3
[9] yaml_2.3.5 utf8_1.2.2 rlang_1.0.6 jquerylib_0.1.4
[13] later_1.2.0 pillar_1.8.1 glue_1.6.2 withr_2.5.0
[17] DBI_1.1.3 plyr_1.8.7 lifecycle_1.0.3 stringr_1.4.0
[21] munsell_0.5.0 gtable_0.3.1 evaluate_0.16 labeling_0.4.2
[25] knitr_1.39 callr_3.7.1 fastmap_1.1.0 httpuv_1.6.5
[29] ps_1.7.1 fansi_1.0.3 highr_0.9 Rcpp_1.0.9
[33] promises_1.2.0.1 scales_1.2.1 cachem_1.0.6 jsonlite_1.8.0
[37] farver_2.1.1 fs_1.5.2 gridExtra_2.3 digest_0.6.30
[41] stringi_1.7.8 processx_3.7.0 getPass_0.2-2 rprojroot_2.0.3
[45] cli_3.4.1 tools_4.0.5 magrittr_2.0.3 sass_0.4.2
[49] patchwork_1.1.2 tibble_3.1.8 whisker_0.4 pkgconfig_2.0.3
[53] ellipsis_0.3.2 assertthat_0.2.1 rmarkdown_2.15 httr_1.4.3
[57] rstudioapi_0.13 R6_2.5.1 git2r_0.30.1 compiler_4.0.5